

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Recently, aging has become a familiar phenomenon around the world. Aging societies are faced with various chronic diseases, such as Alzheimer's disease (AD), obesity, and type 2 diabetes (T2DM). Globally, an estimated 422 million adults over the age of 18 years suffered with diabetes in 2014, and 5.3 million Americans of all ages were diagnosed with AD in 2015.1 According to the 2012 Korean Epidemiological Survey of Dementia, around 530 thousands Koreans older than 65 years were diagnosed with AD, with an estimated two-fold increase therein every 20 years. Interestingly, obesity, T2DM, and AD are reported to be related with each other.2
AD is a degenerative brain disease and the most common cause of dementia. AD is characterized by extracellular amyloid beta (Aβ) plaques and intraneuronal deposits of neurofibrillary tangles (NFTs). NFTs are constituted by hyperphosphorylated tau proteins, whereas Aβ plaques are insoluble and aggregated form of Aβ peptide.3 Until now, therapeutics for AD has been focused mostly on Aβ, with little success. Development of AD treatment could potentially benefit from seeking to consider the relationships among AD, obesity, and T2DM.
According to the Mayo Clinic Alzheimer Disease Patient Registry, 80% of AD patients show impairment in glucose tolerance or have diabetes.4 Epidemiological studies have demonstrated that T2DM induces cognitive impairment and that patients with T2DM are more likely to be diagnosed with dementia by 1.5- to 2-fold.5 Also, increasing evidence has shed light on cellular insulin resistance or insulin insufficiency in the brains of AD patients, including those without having diabetes, thus AD is often referred to as “type 3 diabetes”.6
Obesity is generally defined by a body mass index (BMI) over 30 kg/m2 (25 kg/m2 in Korea) and is usually caused by physical inactivity and westernized diet habits. Obesity is an important risk factor for various diseases, ranging from metabolic diseases to neurodegenerative diseases. As obesity causes insulin resistance, it is one of the major risk factor for T2DM. After years of hyperinsulinemia, insulin secretory function in the pancreas can falter, causing patients to suffer from relative hypoinsulinemia, which leads to hyperglycemia.7
A large number of research has proven that obesity in middle age individuals can be an index of mild cognitive impairment at later years.8 Even when controlling for aging, studies have shown a negative correlation between BMI and global cognitive performance.9 T2DM has also been linked to reduced cognitive function and an increased risk for developing dementia, including AD.10
AD, obesity, and T2DM share similar demographic profiles, risk factors, and clinical and biochemical features.11 These conditions are associated with chronic inflammation, severe oxidative stress, and impairment in insulin signaling and energy metabolism.12 Interestingly, people with diets high in cholesterol, saturated fats, and total calories have been found to be at greater risk for AD than people who regularly eat fiber, vegetables, and fruits.13 Animal studies have also demonstrated that a high-fat diet causes AD pathology, including accumulation of Aβ and phosphorylated tau proteins, as well as cognitive impairment.1415
Given the relationship among obesity, T2DM, and AD, understanding the mechanistic links among them is essential to developing effective strategies for AD prevention and treatment. Until now, inflammation has been thought to link obesity, T2DM, and AD.116 Considering AD as a metabolic disease, however, we suggest that metabolic alterations could be the primary mechanistic links among these diseases. Therefore, the specific goal of this review paper was to investigate proper therapeutic targets for treating AD from our point of view that AD is a metabolic disease.
ALZHEIMER'S DISEASE: METABOLIC ALTERATIONS IN THE BRAIN
AD is a degenerative disease that impairs cognition and memory and is a leading cause of dementia. Alois Alzheimer, for whom AD is named, described the pathology of the disease as an extensive distribution of neuronal tangles and amyloid pl-aques in the brain, which are considered hallmarks of the disease.3 Aβ accumulation is thought to initiate AD by destroying synapses causing neuron loss, a process known as amyloid hypothesis. Amyloid hypothesis has become the dominant model of AD pathogenesis and has guided the development of potential treatments for AD.17
However, clinical studies have documented several inconsistencies in AD pathology. Around 99.6% of drug candidates targeting amyloid pathways, such as beta secretase inhibitors, gamma secretase inhibitors, and Aβ itself, have failed.18 In addition, a weak correlation between Aβ deposition and cognitive decline have been reported.19 For example, Aβ deposits are observed in the medial prefrontal cortex at early stages of AD. Then, these deposits expand into the medial temporal lobe, including the hippocampus. However, cognitive function begins to decline in an opposite way, starting from the medial temporal lobe to the medial prefrontal cortex.20
Recently, early selective atrophy and alteration of glucose metabolism were discovered in the medial temporal lobe of patients with AD using imaging tools.21 In addition, in Alzheimer's disease, degeneration of the basal forebrain cholinergic neurons and a decrease in brain glucose metabolism are characteristically observed.22 With two functional brain imaging techniques, the [18F] 2-fluoro-2-deoxy-D-glucose positron emission tomography and functional magnetic resonance imaging, alterations of glucose metabolism in the brain have begun to receive attention as a feasible cause for AD.23 Interestingly, patients with AD have been found to display increased lactate levels in cerebrospinal fluid (CSF), resulting from metabolic dysfunction;24 however, this type of metabolic alteration has only now been considered as a characteristic of AD.
Aging plays critical roles in neurodegenerative disease and metabolism and is an important cause of AD. As an individual's metabolism gets weaker with age, metabolic alterations are feasible consequences of aging and potential causes of AD. Indeed, imaging studies have shown that a person aged over 70 years could have presymptomatic AD if individuals exhibit dysregulation in glucose metabolism, even though they may show normal cognition at present.25 However, amyloid hypothesis considers AD as a disease determined exclusively by the genomic instability not by metabolic alterations.26
There are two types of cells in the brain, neuron and glial cells, and they are reported to be related with many components of metabolism, called neuron-astrocyte shuttle hypothesis.27 Both cells make use of glucose as a primary energy source, although they metabolize glucose in different ways. In astrocytes, most glucose is metabolized to lactate anaerobically, and then, lactate is released into the extracellular space. In neurons, pyruvate derived from glucose or lactate is metabolized under aerobic conditions, and the energy is produced mainly by oxidative phosphorylation in the mitochondria. Because neurons do not have enough activated glycolysis prompting enzymes, neurons are inclined to not process glucose through glycolysis.28
Due to aging, mitochondria can malfunction, resulting in excessive increases in oxidative stress, as well as oxidative phosphorylation, in the brain.29 Increased mitochondrial oxidative activity demands more energy substrates to produce energy, such that there is an energy shortage in the whole body even though the other cells that have not experienced increased oxidative activity including the brain suffer energy shortage.30 Thereby, some neurons are unable to occupy energy substrates and produce enough energy. Lacking energy, these neurons cannot survive and cause dementia.
Metabolic diseases, such as T2DM and obesity, are reported to be related with AD, meaning that AD is a kind of metabolic disease (Fig. 1). Therefore, metabolic alteration could be potential cause of AD, such that various substances (insulin, adiponectin, and antioxidants) that are influenced by metabolic alterations could be therapeutic targets for AD.
ALZHEIMER'S DISEASE: BRAIN INSULIN RESISTANCE
Considering the prevalences with which T2DM and AD occur, the idea that patients with T2DM could be at higher risk for AD has garnered greater acceptance. In addition, patients with T2DM have been found to be at a two-fold higher risk for AD and to be diagnosed with mild cognitive impairment in a comparably shorter time.31 Hyperglycemia has also been found to accelerate cognitive decline.32 As the brain is likely to encounter defective insulin signaling with increasing age,33 researchers have begun to study mechanistic links between these diseases; the most promising link has been insulin.34
Insulin is a major anabolic hormone that is secreted from pancreas beta cells in response to high glucose levels. Insulin signals by binding to insulin receptors, thereby activating insulin receptor substrate 1, extracellular signal-related kinase/mitogen activated protein kinase, and PI3 kinase/Akt pathways. Insulin also inhibits glycogen synthase kinase-3 (GSK-3).35 Through these signals, insulin maintains glucose homeostasis by regulating glucose production from the liver and glucose consumption by muscle and adipose tissue via GLUT4 translocation. Accordingly, impairment of insulin secretion or function is considered a primary cause of diabetes.36
Insulin resistance refers to the reduced ability of insulin on target tissues, including liver, muscle, and fat tissues. Specifically, insulin receptors' ability to transmit downstream signals or the ability of insulin to activate insulin receptors are reduced in insulin responsive tissues, and these phenomena have been considered as a hallmark feature of T2DM.37 While the vital role of insulin in the periphery is well recognized and has been extensively studied, the function of insulin in the central nervous system has only recently been studied.
Due to the large size of insulin peptide, it was thought that insulin is unable to cross the brain blood barrier (BBB), such that the brain was regarded as an insulin-independent organ.38 However, several studies have revealed that insulin receptors are abundantly distributed throughout the brain, proposing some mechanisms to explain the existence of insulin in the brain.39 Currently, insulin has been shown to be transported across the BBB through a carrier-mediated, saturable, and temperature- sensitive active process.40
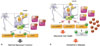
All tissues and cell types, including neurons, have conserved insulin-signaling pathways.41 As shown in Fig. 2A, insulin participates in neuromodulation to regulate the concentrations of neurotransmitters, such as acetylcholine,42 repair, neuronal differentiation, proliferation, regeneration, and inhibition of neuronal apoptosis.43 Through downstream cascade, insulin mediates long-term potentiation, learning, and memory processes.4445 Insulin signaling in specific brain regions, such as the limbic system and hypothalamus, has been found to be necessary for cognitive function.46
Interestingly, patients with diabetes also show insulin resistance in the brain.47 As Fig. 2B describes, reduced insulin action causes AD like alterations in the brain.48 In turn, Aβ oligomers initiate to remove insulin receptors from plasma membranes.49 Following these discoveries, insulin has been measured in postmortem brain tissue from AD patients, showing relatively low levels of insulin.50 Previously, we demonstrated that long-term high fat diet induces T2DM with insulin resistance both in the body and the brain, as well as AD pathologies, such as cognitive defects, accumulation of Aβ, and hyperphosphorylated tau in ICR mice.51
Insulin affects Aβ accumulation and the phosphorylation of tau as well. Insulin-degrading enzyme (IDE) is known to degrade excess insulin and other substrates, such as Aβ (Fig. 2). IDE knockout mice display accumulation of Aβ, hyperinsulinemia, and hyperglycemia.52 As insulin levels are raised, IDE expression is activated to prevent the long-term activation of insulin. However, when IDE is occupied with excess insulin, IDE cannot degrade Aβ, leading to the formation of senile plaques.53
GSK-3 has been most extensively studied as a kinase for tau, although it is also involved in Aβ production.54 GSK-3 is a multifunctional serine/threonine kinase that is affected by both the phosphorylation and aggregation of tau. One study has revealed that inhibition of GSK-3 improves learning and memory and decreases phosphorylation of tau in AD transgenic mouse models.55 We also demonstrated that GSK-3 is inhibited in the brain of mice with AD pathologies.51
Notably, intranasal administration of insulin has been found to help normal adults with improved memory by maintaining their serum insulin and glucose levels.56 Furthermore, intranasal insulin treatment has been shown to enhance cognitive performance in early AD patients,57 suggesting that insulin could be therapeutic target for AD.
ALZHEIMER'S DISEASE: REDUCED LEVELS OF ADIPONECTIN INDUCED BY OBESITY
The incidence of obesity is steadily rising throughout the world.58 Obesity can have damaging effects on many organ systems,59 because many changes induced by obesity are related to metabolic syndrome, characterized by excess weight, high triglyceride levels, and insulin resistance. Moreover, the incidences of cognitive decline and AD are increased with obesity.60
In normal adipose tissue, adipocytes secrete bioactive molecules, termed adipokines, including leptin, and adiponectin.61 Adipokines regulate several important physiological functions: for example, appetite, satiety, energy expenditure, insulin sensitivity and secretion, glucose and lipid metabolism, fat distribution, hemostasis, blood pressure, neuroendocrine regulation, and function of the immune system.62
In obesity, adipocytes become bigger, and hypertrophic adipocytes lead to the development of a pro-inflammatory environment. Hypertrophic adipocytes are often necrotic, and infiltration of macrophages is increased. Activated macrophages gather around necrotic adipocytes, where they secrete pro-inflammatory cytokines, such as TNF-α, IL-1β. This condition has systemic effects and is linked with the initiation of T2DM pathology.63
Among adipokines, adiponectin has beneficial effects on patients with T2DM and has been used in T2DM treatment due to its role of increasing insulin sensitivity.64 Adiponectin has been shown to enhance insulin sensitivity and several studies have reported the relationship between an increased prevalence of diabetes and decreased levels of adiponectin.65 Overexpression of adiponectin prevents high-fat diet-induced obesity in rodents, and genetically eliminating adiponectin in obese mice facilitates increased fat in the liver.66 Adiponectin reduces hepatic lipogenesis and increases β-oxidation through adiponectin receptor 1 (adipoR1)-mediated activation of adenosine monophosphate kinase (AMPK) and peroxisome proliferator-activated receptor α (PPAR α).67
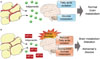
While many researchers have maintained that adiponectin cannot cross the BBB, adiponectin is observed in CSF, and receptors for adiponectin are observed in the brain, suggesting the existence of adiponectin in the brain.68 As shown in Fig. 3A, adiponectin functions in the brain like insulin. As adiponectin receptors are expressed widely in the brain, including the hypothalamus, brainstem, cortex, and pituitary gland, adiponectin signaling pathways are activated in the brain and regulate energy expenditure, food intake, inflammation, cell death, and protection.69
Recently, the relationship between adiponectin and AD has begun to be studied. Some researchers have found a positive correlation between plasma and CSF adiponectin levels. Patients with mild cognitive impairment or AD have significantly higher plasma and CSF adiponectin levels than aged controls.70 However, some researchers found no significant differences in the plasma adiponectin levels between AD patients and non-AD controls. The exact relationship between adiponectin levels and the occurrence of AD remains unclear, although ongoing research suggests a complex association between AD and adiponectin.71
Because AMPK acts as a general energy sensor in the CNS,72 inhibition of adiponectin could affect brain metabolism (Fig. 3B). The local up-regulation of hypoxia inducible factor-1α (HIF-1α) in adipocytes in obese individuals is induced by hypoxic environment due to hypertrophic adipocytes.73 A study found that HIF-1α inhibits production of adiponectin. When adipocyte-specific HIF-1α knock-out mice were fed a high-fat diet for 7 weeks, they displayed high levels of adiponectin mRNA; meanwhile, control mice showed significantly low levels of adiponectin mRNA.74 These conditions could induce metabolic alteration in the brain and initiate pathology of AD.
As we mentioned above, a high fat diet can induce AD pathologies.1415 To find a connection among HIF-1α, adiponectin, and AD, we fed a 60% high fat diet to 8 weeks old ICR mice for 24 weeks. These obese mice displayed cognitive decline and accumulation of Aβ and phosphorylated tau in the brain (under submission). We also revealed that 24 weeks of a high fat-diet induces reductions in the amounts of adiponectin and adipoR1, as well as increase in HIF-1α, in the brain, especially the cortex (Fig. 4).
Even though glucose and lactate are known as major energy sources in the brain, they are not enough to satisfy the energetic demands of the brain. A study revealed that the amount of glucose consumption and the levels of oxygen utilization in the brain are not matched.75 In terms of lactate, large amounts of lactate are unable to generate energy, because cells remove lactate very well and tissues that generate lactate consume most of it.76
After these discoveries, researchers found that fatty acids can enter the brain and mitochondria can oxidize them in the brain.77 Even though some studies have revealed that fatty acids are unable to be used as fuels for brain energy, mitochondrial oxidation of fatty acids provide energy to the brain, maximally 20% of its total energy.78 Furthermore, fatty acids play important roles in glucose homeostasis.79 Fatty acid metabolism is related with adiponectin, so that alterations of adiponectin could induce brain metabolism alteration and AD. Furthermore, adiponectin could be a potential therapeutic target for AD.
POSSIBLE CAUSE OF ALZHEIMER'S DISEASE: REDUCED ANTIOXIDANT DEFENSE MECHANISMS INDUCED BY VARIOUS METABOLIC ALTERATIONS
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are defined as reactive chemicals that have the capacity to donate electrons (e-) to various molecules.80 In cells, ROS/RNS are produced through normal metabolism under aerobic physiological conditions.81 These species are generated under not only normal but also pathological conditions and result in oxidative stress, including nitrogen-based free radical species, as well as superoxide free radicals and hydrogen peroxide.82
Physiological levels of ROS/RNS play an important role in maintaining homeostasis and regulating signal transduction involved in proliferation and survival.83 Cellular antioxidant defense mechanisms regulate ROS/RNS production to maintain proper amounts thereof in normal environments. However, when ROS/RNS formation is dysregulated, oxidative stress disrupts cellular function and damages cells.8284
Research indicates that Aβ causes oxidative stress. Aβ can form complexes with copper (Cu), generating hydrogen peroxide via Cu2+reduction.85 Aβ-induced oxidative stress disturbs cellular signaling and initiates a phosphorylation response, resulting in activated JNK, p38 MAPK, and hyperphosphorylated tau proteins in AD postmortem brains.86 Furthermore, since Aβ is generated from the cleavage of amyloid precursor proteins, located in trans membrane, it inserts into the bilayer of cells and initiates lipid peroxidation and a series of reactions, producing ROS.87
ROS levels are increased in obesity.88 Many studies have demonstrated that reduced insulin signals and adipokines dysregulation in T2DM patients leads to increased ROS production.89 Furthermore, high glucose levels are able to cause oxidative stress by various mechanisms, including glucose autoxidation, polyol pathway, advanced glycation end products formation, and PKCβ1/2 kinase.90 Increased free-radicals are able to attack unsaturated fatty acids oxidation in physiological systems. Obesity and T2DM patients have increased amounts of some by-products of lipid peroxidation, such as conjugated dienes and malondialdehyde.91
There are essential enzymes in antioxidant defense systems, such as glutathione reductase, glutathione S-transferase, and glutathione disulfide.92 T2DM patients show high oxidative da-mage and reduced antioxidant defenses mechanisms.93 Oxidative stress induced by various conditions, including abnormal superoxide dismutase and leaking mitochondria, is known to contribute to AD.94
In normal conditions, ROS is detoxified by producing succinate 95 via the stabilization of HIF-1α.96 When oxygen supply is adequate, mitochondrial lactate dehydrogenase in astrocytes is used to produce pyruvate and help in the synthesis of adenosine triphosphate. However, under ROS stress, these detoxification processes are unable to be effective. The process may lead towards lipid synthesis.97 Abnormal accumulation of lipid is commonly observed in patients with AD.98
Considering the noted relationships among obesity, T2DM, and AD, it is possible that increased ROS induced by metabolic alterations can cause AD pathologies (Fig. 5). There are several studies that demonstrate the effects of antioxidants on AD treatment. Various vitamins, such as Vitamin A, B, C, and K, are proven to be effective in AD treatment.99 We have also demonstrated the effect of agmatine, a polyamine that works as an antioxidant, on mice displaying AD like alterations induced by long term high-fat diet and rats with streptozotocin-induced AD.51100 Oxidative stress is one of the most important factors involved in the development and progression of AD. Using antioxidants is a feasible approach that can help treat AD.
CONCLUSION
Conventional approaches to developing therapeutics for AD have focused on the Aβ hypothesis, which insists that Aβ causes AD pathologies. These approaches, however, have failed to treat or prevent AD. Through this review, we would like to suggest that metabolites, such as insulin, adiponectin, and ROS, could be therapeutic targets for developing effective treatments for AD, considering that metabolic alterations may be the primary causes of the disease.


 XML Download
XML Download