

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Spinal muscular atrophy (SMA) is a hereditary disease of the anterior horn cell in the spinal cord, which results in progressive proximal muscle weakness. This autosomal recessive disease has a prevalence of approximately 1 in 10000 newborns. In the 1990s, the Survival Motor Neuron (SMN) gene was revealed to be a key gene involved in SMA. The most common form of SMA is caused by homozygous deletions in the Survival Motor Neuron 1 (SMN1) gene accounting for about 95% of reported cases.1 Patients with SMA are typically classified into four subtypes according to age of onset and degree of severity: type I–IV.2
Owing to its clinical similarity with congenital myopathy, clinicians have some difficulties in quick and direct diagnosis of SMA. Conventionally, electrophysiological tests, such as electromyography, and pathologic study by muscle biopsy are conducted as a primary study in patients with proximal dominant symmetric progressive weakness. It is now advisable to analyze the SMN1 deletion along with electrophysiological and pathologic study in the initial assessment of typical SMA-like patients, since genetic diagnostic test can be easily performed and offers accurate diagnosis, with a sensitivity of up to 98%.23
With improvements in genetic analysis and novel therapeutic approaches, such as gene therapy and molecular therapy, the diagnostic approach for patients with SMA is significantly important. We investigated the clinical and pathologic characteristics of SMA cases confirmed by a genetic test in our hospital.
This study is a single center oriented, retrospective, and descriptive study. We first reviewed 42 patients referred to Gangnam Severance Hospital in Seoul from September 2007 to February 2016 who had received SMN1 genetic test. Genetic analysis of the deletion of exon 7 of the SMN1 gene on chromosome 5q13 was performed by multiplex ligation-dependent probe amplification. After exclusion of 14 patients who showed no deletion of exon 7 or had little information in their medical records, we finally included 28 patients and reviewed their clinical and pathological features from the medical records. Type of SMA was classified based on the age of onset as type I with onset before 6 months, type II from 6 to 18 months, and type III after 18 months.
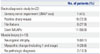
Of the total 28 patients, there were 15 male and 13 female patients. Seven out of 28 patients had familial history with similar symptoms. Clinical types of SMA were distributed as 3 patients with type I, 14 patients with type II, and 11 patients with type III. All patients had proximal dominant symmetric weakness regardless of the type of SMA. Eight patients showed dysphagia and 2 patients showed dysarthria. Two patients with type II and 1 patient with type III showed tongue fasciculation associated with dysarthria and ventilator care. Ventilator support was needed in 15 patients, including all 3 patients with type I, 8 patients with type II, and 4 patients with type III. Twenty-five patients had scoliosis, and 8 of these patients, including 1 patient with type I, underwent a surgical treatment for scoliosis with an improvement in clinical outcomes (Table 1).
Twenty-two patients underwent an electrodiagnostic study (EDS). Among them, 15 patients had both an EDS and muscle biopsy. Based on the clinical features together with EDS and muscle biopsy results, 9 patients were diagnosed with SMA, while the other 6 patients were diagnosed with myopathy (Table 2). These 6 patients were later found to be misdiagnosed with myopathy (Table 3). Among them, we noted that EDS fi-ndings were interpreted as myopathy in 4 patients and neuropathy in 1 patient. One patient diagnosed at the age of 3 did not have an EDS. The clinical pattern of symmetric proximal dominant limb weakness was the same in all 6 patients. Therefore, the EDS findings and clinical patterns together could have generated the confusion. These 6 patients were later confirmed to have SMA after SMN1 gene mutation analysis. Of these patients who were misdiagnosed with myopathy before the genetic study, it took as long as 11 years for these patients to achieve the correct diagnosis of SMA through a genetic study.
The spectrum of clinical severity of SMA ranges from infantile paralysis causing premature death to mild motor weakness and normal life expectancy. Generally, patients with SMA type I hardly survive until 48 months; however, according to recent reports, there have been some studies showing a decreasing mortality in SMA type I patients.4 In our study, three of the SMA type I patients had survived through the age of 20 years at the last follow up, even though all of them required the use of a ventilator. These patients were all conservatively well-managed from their infantile period through routine follow up in an outpatient clinic. Supportive care such as this including respiratory support and surgical correction of scoliosis are very important for better clinical outcomes. Currently, there are no curative treatments that effectively alter the natural history of the disease. The goal of supportive care is to lessen the disease burden through managing symptoms and preventing complications. Adequate nutritional intake with physical therapy is important, and proper respiratory support is crucial since respiratory failure is a serious problem in patients with SMA type I and II. Noninvasive positive pressure ventilation may provide improved quality and life expectancy.
The majority of patients who survive childhood develop progressive scoliosis, which worsens pulmonary function.5 However, few studies have demonstrated significant improvement after surgical correction of scoliosis.6 In our study, patients who had surgery to correct scoliosis showed improvements in pain relief, muscle strength during sitting or walking, frequency of respiratory infection, and some showed increase in vital capacity. These findings are similar to recent studies showing that surgical trunk alignment correction is essential to improve sitting and standing tolerance, as well as pulmonary function, in SMA patients.7
In our study, weakness due to SMA preferentially involved proximal muscles, in particular the lower extremities were the first involved. The clinical course of SMA can sometimes be confused with congenital myopathy. This clinical similarity leads clinicians to a misdiagnosis or delayed diagnosis for some patients.
Among the 22 patients in our study who underwent an EDS, many showed spontaneous activities, while some patients also had myopathic findings (Table 2). Among them, 15 patients had a muscle biopsy prior to genetic analysis. It is interesting that only 9 patients were diagnosed with neurogenic atrophy compatible to SMA, while 6 patients showed normal findings or had been misdiagnosed with myopathy. EMG results sh-owing polyphasic motor unit action potentials together with clinical similarity could have led clinicians to misinterpret the pathologic findings of muscle biopsy. Also, the serum CK level did not show meaningful differences to help distinguish between SMA and myopathy (Table 3). These days, molecular genetic analysis for the SMN1 gene deletion is becoming the gold standard diagnostic modality for correct diagnosis. Genetic analysis of the absence of SMN1 exon 7 confirms the diagnosis of SMA with 95% sensitivity and nearly 100% specificity.8 For this reason, we emphasize that the best approach for SMA diagnosis uses genetic analysis to detect a homozygous SMN1 deletion, which is a simple and non-invasive test.9
The limitations of this study are that only a small relevant population was included. Also, even though subgrouping of SMA is currently still done on a clinical basis, it would have been a better review with analysis of SMN2 gene copy number of patients. We could not determine the copy number of SMN2 due to the lack of data. These days, the association between SMN2 gene copy number and clinical phenotype in patients with SMA is well known, and its possible effect as a phenotype modifier is being examined.1011 Even though SMN2 produces less of the full length protein transcript than SMN1, it is known the number of SMN2 copies affects the phenotype of SMA.12
This study reviewed the clinical phenotype and genetic distribution in patients with SMA who were diagnosed as having the SMN1 deletion by multiplex ligation-dependent probe amplification in Korea. Since upcoming novel gene therapy requires a potential target for appropriate treatment, having proper knowledge of the genetic profile of patients in the Korean database is important. Moreover, EDS and muscle biopsy conducted before genetic analysis led to confusion and made the accurate diagnosis of SMA rather difficult, while the correct diagnosis was achieved with genetic mutational analysis. Therefore, we emphasize that genetic analysis should be done early in the diagnostic process for patients with symmetric and proximal dominant weakness.


 XML Download
XML Download