

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Traumatic brain injury (TBI) is a leading cause of mortality and morbidity worldwide, particularly in individuals younger than 45 years of age.12 Currently available treatments cannot reverse the primary injury of trauma; therefore, most treatment strategies focus on limiting injuries secondary to TBI and improving outcomes after TBI. However, current treatment options for TBI are limited, and cell therapy is a promising therapy under investigation.
Angiogenesis is a critical process for repairing TBI-induced brain damage, and the process provides the neurovascular substrates necessary for neuronal remodeling.34 Endothelial progenitor cells (EPCs) are bone marrow-derived stem cells that play an essential role in the process of vessel repair upon injury.5 Previous studies have shown that the number of circulating EPCs is first suppressed in the acute phase of TBI and then rapidly increased. These dynamic changes are positively correlated with clinical outcomes in TBI patients.67 Animal studies demonstrated that venous or intracerebroventricular infusion of ex vivo expanded endothelial colony-forming cells accelerates angiogenesis, repairs the blood-brain barrier, and improves cerebral function after TBI in mice.89 However, a hostile microenvironment poses a significant challenge to the survival of transplanted cells. Only a small fraction (1–3%) of transplanted EPCs survive 28 days after transplantation, a fact that greatly limits their potential therapeutic effects.1011
The accelerated death of grafted cells may result from the production of reactive oxygen species (ROS) after ischemic reperfusion injury and host inflammatory response mediators.12 Thus, providing an antioxidative environment may have a protective effect on endogenous cells or transplanted cells. To protect itself from oxidative damage, the brain produces numerous antioxidants. Superoxide dismutase (SOD) is the major endogenous antioxidant enzyme that provides first line protection against oxidative stress.2 On the other hand, lipid peroxidation is one of the principal outcomes of oxidative stress-mediated tissue injuries; malonyldialdehyde (MDA), a metabolite of polyunsaturated fatty acids via peroxidation, is often used as a surrogate marker for oxidative stress.13
Although the roles of SOD and MDA after TBI have been studied, the association between levels of SOD or MDA and circulating EPCs after TBI has not been investigated. The present study is an initial, observational study that is aimed to assess if a correlation between oxidative stress and circulating EPCs can be established.
Go to : 
MATERIALS AND METHODS
Patients
Twenty patients with mild, closed TBI [the severity of TBI based on the Glasgow Coma Scale (GCS) was 13 to 15] were included in this study. Patients with complex trauma involving the body trunk and limbs, hematologic disorders, cancer, and infection were excluded from this study. Twenty age- and sex-matched healthy subjects were recruited for the study as control group. Blood samples were collected 24 hours and 2, 3, 4, and 7 days after injury. This study protocol was approved by the Ethical Committee of Shanxi Medical University First Affiliated Hospital. Signed informed consent forms were collected from all patients or guardians before enrollment and blood samples were collected.
EPCs isolation and measurement
Blood samples (2 mL) from patients were diluted with phosphate-buffered saline (pH 7.2) and centrifuged with Ficoll-Hypaque for mononuclear cell (MNC) preparation. Circulating EPCs were measured from isolated MNCs by flow cytometry. Cells that were double-labeled with antibodies against CD34 and CD133 were considered as EPCs, as described previously.6
Oxidative activity analysis
The plasma levels of SOD and MDA were measured using the SOD activity colorimetric assay kit and the lipid peroxidation (MDA) assay kit according to the procedure supplied by the manufacturer (Abcam, Cambridge, MA, USA).
Statistical analysis
Data are presented as mean±standard deviation unless otherwise noted. The circulating EPCs, SOD, and MDA levels between mild TBI patients and control subjects at the same time point were compared by an independent-samples t-test. For within group comparison, one-way ANOVA with repeated measures was performed. Correlation between circulating EPC number and plasma SOD or MDA levels was detected by Pearson correlation coefficients. A statistical significance was defined at an error probability of <0.05. Data were analyzed using SPSS software (16.0; SPSS Inc., Chicago, IL, USA).
Go to :
RESULTS
Twenty patients with mild TBI (mean age of 51 years; 15 men and 5 women), and twenty healthy controls (mean age of 45 years; 16 men and 4 women) were included in the study. The two groups did not differ statistically in sex (p=1.000) and age (p=0.128). Causes of TBI included vehicle accidents (15 cases), fall from height (2 cases), and physical assaults (3 cases). Computed tomography scan was performed on all patients to determine the types of injuries. Diagnosis at the time of admission included epidural hematoma (2 cases), subdural hematoma (5 cases), intracerebral hematoma (2 cases), contusion/laceration (14 cases), traumatic subarachnoid hemorrhage (8 cases), and skull fracture (6 cases). No patient underwent surgery, as surgery is not necessary in treating mild TBI. Demographics and clinical characteristics of the patients are summarized in Table 1. Twenty age- and sex-matched volunteers were included in the study as the control group.
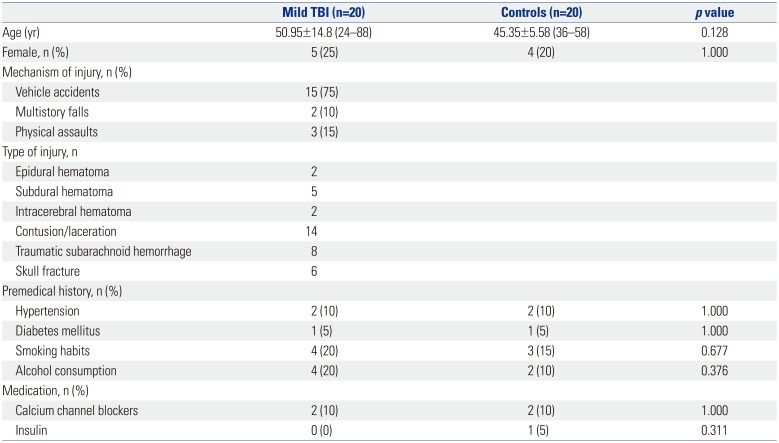
Table 1
Demographics and Clinical Characteristics of Mild TBI Patients and Healthy Controls
![]()
The average circulating EPC number in the control group remained stable throughout the 7-day monitoring period. In contrast, the average circulating EPC number in mild TBI patients decreased initially and was the lowest at day-2 post-TBI, measuring 50.21±6.17, which was significantly lower than the control group (75.32±5.94, p<0.001). However, it increased progressively and measured 132.27±15.41 at day-7 post-TBI, significantly higher than the control group (74.34±5.65, p<0.001). Of note, the circulating EPC numbers from day-2 to day-7 were significantly different when compared with day-1 (p=0.003, day-1 vs. day-2; p<0.001, day-1 vs. day-3 to day-7) (Fig. 1).
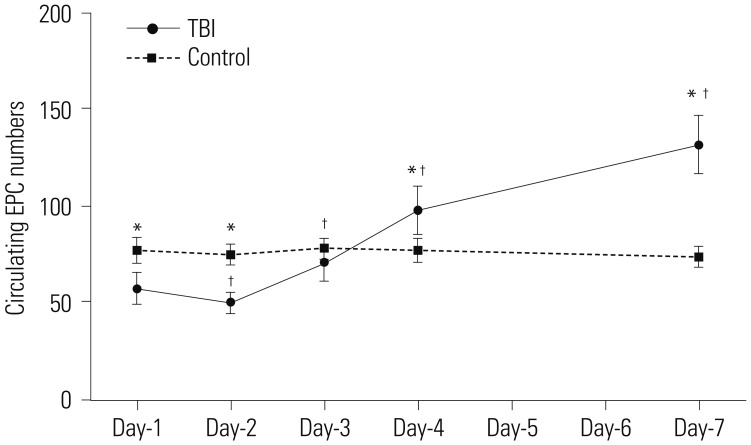 | Fig. 1Dynamic change in number of circulating EPCs post mild TBI. Circulating EPC numbers in peripheral blood samples were measured at day-1, day-2, day-3, day-4, and day-7 after TBI during the follow-up period of 7 days. For controls, circulating EPC numbers from 20 healthy subjects were measured at the same intervals (*p<0.01 vs. control; †p<0.01 vs. day-1). EPC, endothelial progenitor cell; TBI, traumatic brain injury.
|
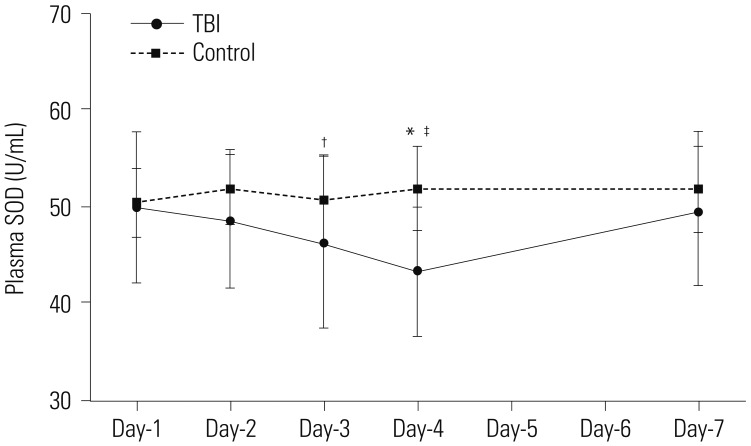
We next examined plasma levels of SOD, an antioxidant; and MDA, a marker for oxidative stress. The plasma levels of SOD and MDA remained stable throughout the monitored period in the control group. The plasma levels of SOD in TBI patients were no different from the control group at day-1 post-TBI, but decreased progressively, and were lowest at day-4 post-TBI, measuring 43.44±6.58 U/mL, which was significantly lower than those in the control group (51.92±4.41 U/mL, p<0.001) (Fig. 2). Plasma levels of SOD recovered gradually thereafter and were not different from those with the control group at day-7 post-TBI. Furthermore, the plasma levels of SOD at day-3 and day-4 were significantly different than those at day-1 (p=0.352, day-1 vs. day-2; p=0.044, day-1 vs. day-3; p=0.001, day-1 vs. day-4; p=0.803, day-1 vs. day-7).
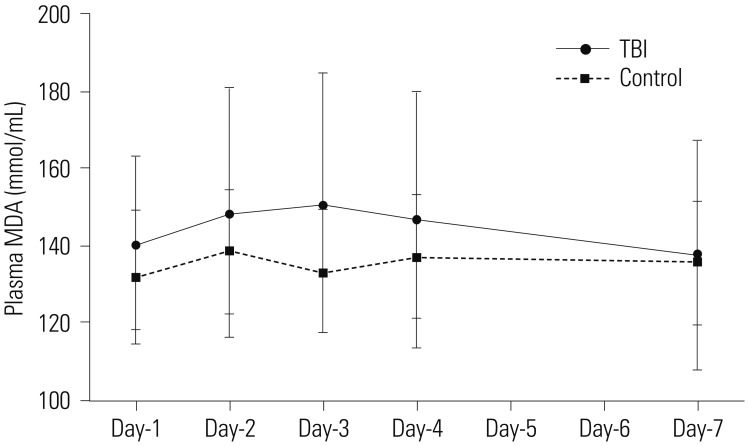
In contrast, the plasma levels of MDA in TBI patients were not different from those in the control group at all-time points assessed, and no statistical difference in the plasma levels of MDA were detected between day-1 and subsequent days (Fig. 3).
To examine the potential correlation between circulating EPC number and markers of oxidative activity, we tested whether circulating EPC number is linearly correlated with either plasma SOD or MDA levels using bivariate correlation tests. For each post-TBI day, circulating EPC numbers from each TBI patient were plotted against corresponding SOD and MDA levels. No linear correlations were detected for day-1 through day-6 post-TBI. However, at day-7 post-TBI, the number of circulating EPCs in TBI patients displayed a weak to moderate positive correlation with plasma levels of SOD (r=0.472, p<0.05), while exhibiting a weak to moderate negative correlation with plasma levels of MDA (r=-0.487, p<0.05) (Fig. 4).
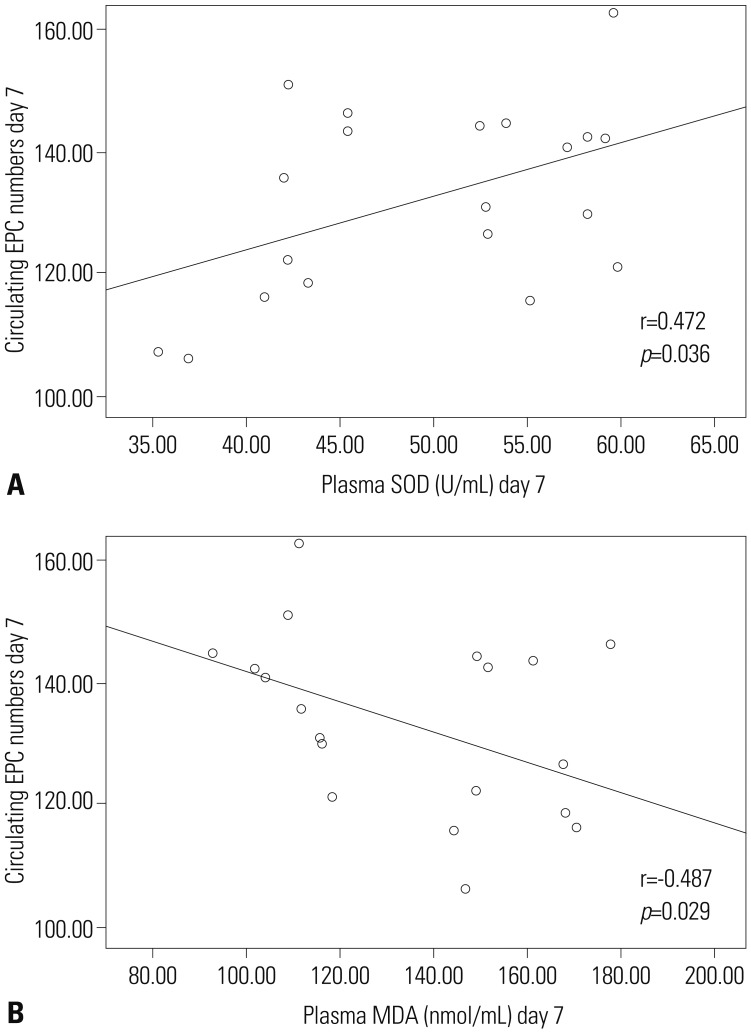 | Fig. 4Correlation analyses between the number of circulating EPCs and SOD or MDA. Circulating EPCs were weakly to moderately correlated with SOD levels at day 7 (A) and with MDA levels at day 7 (B) post mild TBI. EPC, endothelial progenitor cell; SOD, superoxide dismutase; MDA, malonyldialdehyde; TBI, traumatic brain injury.
|
Go to :
DISCUSSION
In this observational study of 20 mild TBI patients, we found that reactive changes in circulating numbers of EPC follow a two-phase trail, where they decrease initially and then increase. In the present study, plasma levels of SOD were decreased at day 3 and day 4 when compared with those at day 1, while plasma levels of MDA did not change significantly. At day-7 post-TBI, we noted a weak to moderate correlation between circulating EPC number and plasma levels of SOD and MDA.
The brain is particularly vulnerable to oxidative stress, as its rate of oxygen consumption is high, and additionally, as transition metals and polyunsaturated fatty acids levels are high.2141516 Cerebral hypoxia and ischemia often occur secondary to TBI. Both conditions result from impaired cerebral blood flow auto-regulation, and lead to increased oxidative stress. As an imbalance between the generation of free radicals and antioxidants progresses, the brain is subjected to oxidative stress, which plays a key role in the progression of secondary damage after TBI.17 Oxidative stress can result from activation of pH-dependent calcium channels in cells in an excitotoxic state. Opening of calcium channels deactivates the mitochondria electron transport chain, which leads to an increase in ROS/reactive nitrogen species production, thereby leading to lipid peroxidation and oxidation of proteins and nucleic acids.
SOD is an antioxidant enzyme that catalyzes superoxide radical anions O2-into hydrogen peroxide (H2O2). Therefore, SOD activities reflect the anti-oxidative capacity of the tissue. Lipid peroxidation is one of the principal outcomes of oxidative stress-mediated tissue injuries, and tissue MDA is an indicator of the content and extent of lipid peroxidation.1318 In the present study, we used SOD and MDA as indices for oxidative stress. Consistent with a previous study that found SOD activities decreased at 24 hours after the onset of TBI and during seven days of follow-up,19 we observed that plasma SOD levels decreased after the onset of mild TBI, indicating accelerated degradation of the protein. Increase in MDA is one of the harmful consequences of TBI that rapidly causes cellular injury.20 Previous clinical studies have described that an increase in plasma and CSF levels of MDA as early as 2–3 hours after severe TBI in infants and children, which persists for at least 7 days after injury.21 In the present study, however, we did not observe a significant increase in MDA after TBI when compared with healthy volunteers. Our observation of the dynamic change in the plasma levels of SOD indicated that, in the early stage of mild TBI (day-1–day-4 post-TBI), patients experience a greater degree of oxidative stress. Although the plasma MDA levels in TBI patients were not different from the control group, a weak trend of change was detected. One possibility as to why the plasma MDA levels did not increase significantly is that the patients in this study were all mild TBI patients, whereas more substantial changes in MDA levels are typical in severe TBI patients.22
Consistent with previous studies,67 the present study found that the number of circulating EPC decreased initially and then increased significantly. The circulating EPC number was significantly higher when compared with the control group at day-7 post-TBI. We hypothesized that there is a potential correlation between circulating EPC number and oxidative stress. When we plotted circulating EPC number from each patient against corresponding SOD or MDA levels, we found a weak to moderate correlation between circulating EPC number and plasma levels of SOD (positive) and MDA (negative) at post-TBI. These results suggest that SOD and MDA may play a role in regulating the number of circulating EPCs at the sub-acute stage of mild TBI. The weak to moderate correlation between circulating EPCs and SOD (r=0.472, p=0.036), and MDA (r=-0.487, p=0.029) was significant only at day-7 post-TBI, probably because of the multifactorial etiology of injury and inter-patient variability.
EPCs are capable of migrating to the area of injury, differentiating into mature endothelial cells, and participating in angiogenesis and vasculogenesis.2324 Thus, cell therapy as an experimental treatment option has been used to treat some TBI patients. The efficiency of cell therapy is determined by multiple factors, particularly the survival of donor cells in the hostile environment of a brain lesion.25 Therefore, it is imperative to identify therapies that can protect donor cells in the host microenvironment. Antioxidant enzymes have been found to protect cells against the effects of oxidative stress of ROS after cerebral ischemia and reperfusion. Previous studies found that enhancing endogenous antioxidant expression of cells could promote survival of these grafted cells.25262728 Our finding that plasma levels of SOD and MDA were weakly to moderately correlated with circulating EPCs number at day 7 suggests that this antioxidant strategy might be valid in cell therapy at sub-acute stage of mild TBI.
One limitation of our study is that patients with moderate and severe TBI are not included. TBI is a complex disorder, and circulating EPCs have been found to be correlated with the severity of the injuries as assessed with GCS.7 Severe TBI patients experience higher oxidative bursts and higher plasma levels of inflammatory mediators than moderate TBI patients.22 Further larger studies are required to determine the causal relation between circulating EPCs, the plasma levels of SOD and MDA, and the severity of injuries in TBI patients.
In summary, we report that the number of circulating EPC and the plasma SOD level exhibited dynamic changes in mild TBI patients at early stage. The number of circulating EPC were weakly to moderately correlated with plasma levels of SOD and MDA at day-7 post-TBI. These findings suggest that providing an antioxidative environment may facilitate successful EPCs transplantation. Additional studies are required to further examine this hypothesis.
Go to :
 XML Download
XML Download