

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The liver is a major organ that controls metabolic flux. For studies related to metabolism, liver-specific expression or inhibition of genes for an extended period without any adverse effects could be an invaluable tool. Adenovirus has been traditionally used to express recombinant genes in the liver. Resourceful protocols for the generation and purification of recombinant adenovirus are well-established, and various kits are commercially available.123 Numerous experiments using adenovirus have been published, and most of them provided satisfactory results in terms of gene expression levels necessary to reach their goals. However, adenoviral-mediated gene expression is limited to short periods, generally less than 2–3 weeks, and there are safety issues in transduced animals and humans during the preparation procedures and experiments.1456
Recently, recombinant gene expression using adeno-associated viruses (AAVs) have been used in animal studies, as they can mediate safe, efficient, and stable transduction and maintain expression of the transduced gene for several months.7 AAVs are considered safe, cause no known human disease, and have been used in clinical trials as a method for delivering genes to humans.89 AAVs are nonpathogenic, replication-deficient single-stranded DNA viruses of the parvoviridae family. AAV2 is the serotype that has been used most frequently. Gene expression using AAV2 is restricted to the liver when systemically injected in animals, though at low levels.101112 AAV8 and AAV9 have been subsequently introduced and have higher levels of hepatic expression than AAV2.131415 However, their broad tissue tropism may raise problems when liver-specific expression is necessary,1416 and the reported purification methods for AAV8 and AAV9 are complicated.17 Recently, AAV-DJ has been developed using a gene shuffling method involving various AAV serotypes, and the viral vector is now commercially available.18 AAV-DJ has the advantageous characteristics of both AAV2 and AAV8. AAV-DJ contains heparin-binding domain in its capsid, similar to AAV2, which suggests that a heparin column could be used for purification.19 AAV-DJ has superior transduction efficiency in the liver as well as various established cultured cell lines, which makes AAV-DJ an attractive viral tool for liver-specific gene expression.18 The use of AAV-DJ in animals has not been reported, likely for the reason that there are no established large-scale preparation/purification methods or protocols. Here, we present a simple large preparation method that can generate up to 3×1013 experiment-grade viral particles and demonstrate liver-specific gene expression via systemic injection in mice.
MATERIALS AND METHODS
Preparation of plasmids
Plasmids used included pHelper (Cell Biolabs, San Diego, CA, USA), pDJ (Cell Biolabs), and pAAV-CAG-GFP. pAAV-CAG-GFP was generated using pAAV-CMV-GFP (Cell Biolabs), pAAV-CMV-MCS (Cell Biolabs), and pAAV-CAG-shuttle-WPRE plasmid (Applied Viromics, Fremont, CA, USA). Green fluorescent protein (GFP) was amplified via polymerase chain reaction (PCR) using 5'-GGATCCATGGTGAG CAAGGGCGAG GAGCTG-3' and 5'-AGATCTCTACTT GAGCTCGAGATCTGAG TA-3' as primers and pAAV-CMV-GFP as a template. The amplified PCR product was cut with BamHI and BglII, inserted in BamHI and BglII sites in pAAV-CMV-MCS, and designated as pAAV-CMV-GFP. The GFP fragment of SmaI and BglII from pAAV-CMV-GFP was ligated with a cleaved product of ApaI and BglII from pAAV-CAG-shuttle-WPRE, and the produced plasmid was designated as pAAV-CAG-GFP. The plasmid without GFP was designated as pAAV-CAG-control. The plasmids were transformed, grown in Terrific Broth overnight, and purified using a QIAGENE mega kit (QIAGENE, Valencia, CA, USA). The integrity of two inverted terminal repeats (ITRs) in pAAV is critical for the packaging of AAV. SmaI sites in ITRs and the integrity of ITRs were confirmed by digesting pAAV plasmids with SmaI.
Transfection of QBI-HEK 293A cells
QBI-HEK 293A cells were plated on 150-mm dish at a density of 1.3×106/dish on day 0, and the cells were cultured in DMEM containing 5% fetal calf serum as well as penicillin and streptomycin. A total of 150 150-mm dishes were prepared. The cell density was determined on day 3 and was 80–90% confluent. Plasmids pHelper (40 µg/dish), pDJ (30 µg/dish), and pAAV-CAG-GFP or pAAV-CAG-control (30 µg/dish) were diluted in OPTI-MEM (Thermo Fisher, Waltham, MA, USA). Additionally, 60 µL/dish of polyethylenimine (PEI; Polysciences, Warrington, PA, USA) solution (1 mg/mL) was diluted in OPTI-MEM and incubated for 5 min at room temperature. The diluted DNA was added to the diluted PEI solution and mixed by gentle vortexing. The DNA-PEI complex was incubated for 30 min at room temperature, and the PEI-DNA complex was then added to the cells. The cells were incubated for 48 h before harvesting.
Preparation of cell extracts
The cells and the medium were transferred to tubes, and the pooled cells were pelleted using a centrifuge at 3000× g for 10 min at 4℃. The medium was discarded, and the cell pellets were resuspended in 300 mL of Buffer A (10 mM Tris-Cl, pH 8.0, 0.15 M NaCl) and transferred to 10 50-mL conical tubes, with 30 mL of extracts in each tube. The cells were disrupted by four cycles of freeze in a dry ice-methanol bath/thaw in a water bath of 37℃/vortex for 30 sec. The cell extracts were then centrifuged at 10000× g for 10 min at 4℃, and the supernatant was collected. Benzonase nuclease (Sigma, St. Louis, MO, USA) was added to the cell extracts (50 unit/mL), and the mixture was incubated for 30 min at 37℃. An equal volume of Buffer A (300 mL) was added to the cell extracts. A 10% sodium deoxycholate solution was added to make the final concentration of sodium deoxycholate in the cell extracts 0.5%, and the solution was then incubated at 37℃ for 30 min. The cell extracts were centrifuged at 5000× g for 15 min. The supernatant was collected and passed through a 0.45-µm low protein binding bottle filter.
Purification of virus using heparin column
One 5-mL Heparin column (GE Healthcare, Pittsburgh, PA, USA) was used for cell extracts from the 150 dishes of 150 mm. The Heparin column was equilibrated with Buffer A by running 25 mL of Buffer A at room temperature using a peristaltic pump (the flow rate was 2 mL/min). The cell extract was loaded onto the column at room temperature followed by washing with 10 mL of Buffer A. Next, the column was removed from the peristaltic pump and attached to a fast protein liquid chromatography (FPLC) system (GE Healthcare) at 4℃ and washed again with 25 mL of Buffer B [1 mM MgCl2 and 2.5 mM KCl in 1×phosphate buffered saline (PBS)]. The virus was eluted in a linear gradient from 0% to 100% of Buffer C (1 mM MgCl2, 2.5 mM KCl, 1 M NaCl in 1×PBS) over Buffer B in 50 mL. During the elution step, a single peak was detected via ultraviolet detector when the concentration of Buffer C reached ≈50% of the linear gradient. The single peak was usually generated over four 1-mL fractions, which were collected and desalted using Centriprep (Millipore, Billerica, USA) with 1×PBS, 5% sorbitol. The final volume of the viral solution was less than 3 mL.
Determination of viral titer via quantitative PCR
Vector-specific primers that recognized the sequence in the poly A region were used: 5'-GGTCTCCAACTCCTAATCTCAG-3' and 5'-AAAATCAGAAGGACAGGGAAGG-3'. A set of solutions of pAAV-CAG-GFP plasmid with concentrations of 103–107 copies/µL were prepared using 1:10 dilutions with water and used as a standard. Serial 10-fold dilutions of the purified viral solution were prepared. Using the diluted standard and the viral solutions, quantitative PCR was performed with SYBR Green mix (Thermo Fisher) as described previously.20 The viral titer was determined by comparing CT values of the viral solutions to the standards. Purity of the virus was determined by running the virus on a gel and staining with a SilverQuest silver staining kit (Thermo Fisher).
Expression of GFP in mice
C57BL/6J, 7-week-old male mice were purchased from Jackson Laboratory. The purified AAV-CAG-control and AAV-CAG-GFP viruses were injected into the mice at doses of 1×1011, 3×1011, and 1×1012 viral particles/mouse via the tail vein. Three weeks after injection, one group of mice were terminally anesthetized with pentobarbital (80 mg/kg body weight) and perfused transcardially with 10% formalin (Sigma). Livers were harvested and incubated in 10% formalin overnight, followed by incubation in 20% sucrose in PBS for 6 h and a brief rinsing with PBS. The liver was frozen in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA, USA) and sectioned using a freezing sliding microtome (Leica Biosystems, Buffalo Grove, IL, USA). The slides were examined under a fluorescent microscope. A second group of mice were sacrificed 3 months after injection, and various organs were harvested and examined to determine the expression level of GFP using an IVIS scanner (Perkin Elmer, Waltham, MA, USA). All animal studies were approved by the University of Texas Southwestern Institutional Animal Care and Use Committee.
RESULTS AND DISCUSSION
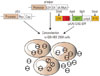
AAV of different serotypes display different capsid proteins and different purification methods. The recently developed AAV-DJ has a hybrid capsid with a heparin-binding domain that may bind to heparin columns strongly enough for purification.19 Instead of using a helper adenovirus, packaging the AAV virus was achieved via co-transfection of a pHelper plasmid that contained E2A, E4, and VA RNA of adenovirus. AAV-DJ virus expressing GFP was produced by the co-transfection of three plasmids (pHelper, pDJ, and pAAV-CAG-GFP) in QBI-HEK 293A cells (Fig. 1). At the end of the 48-h incubation, as the cells produced AAVs, the majority of the cells became round, and some cells detached from the dishes. Using the methods presented in this paper (Fig. 2), approximately 3×1013 viral particles calculated by genome copy number were produced and purified from 150 150-mm dishes. The cell density on the day of transfection was the most important factor in obtaining high yields of virus. The highest yield was achieved at the occurrence of transfection, when the cells were 85–90% confluent. Although transfecting cells at lower densities resulted in a higher transfection efficiency, it ultimately resulted in a lower yield of total viral particles. Transfection of cells at a density higher than 90% reduced transfection efficiency and also resulted in low yields. When AAV-DJ was produced in QBI-HEK 293A cells, ≈40% of the newly assembled virus was secreted into the medium, and 60% of the virus remained in the cells. The protocol presented in this paper was not useful in purifying AAV-DJ from the culture medium, as other proteins in the serum presented in the culture medium also bound to the heparin column, which contaminated the viral fractions from purification. While losing 40% of the total virus produced was not ideal, purifying the virus from a large volume of medium using other methods was more laborious, time consuming, and required the use of expensive reagents. Therefore, we found that using only the cell extracts to obtain pure AAV-DJ was easier, faster, and less expensive.
The purified AAV-CAG-GFP virus was injected into mice via the tail vein (1012 viral particles/mouse), and the expression levels of GFP in various organs were imaged and measured 3 months after injection. The liver was the only organ where GFP was detected despite the use of the ubiquitously active chicken actin promoter (Fig. 3). To determine the titer that expressed GFP protein in 100% of hepatocytes, AAV-CAG-GFP virus was injected at doses of 1×1011, 3×1011, and 1×1012 viral particles/mouse. Three weeks after injection, GFP expression in the liver was examined histologically using a fluorescent microscope. Fig. 4 shows dose-dependent GFP expression in hepatocytes. Approximately 90% of hepatocytes expressed GFP protein at the 3×1011 dose, whereas expression was detected in 100% of the hepatocytes following the 1×1012 dose. We suggest that ≈3×1011 viral particles/mouse be used for general experiments. For secretory proteins, injection doses can be adjusted based on the plasma level of the expressed proteins.



 XML Download
XML Download