

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Essential to a host's immune responses to pathogens is discrimination of non-self molecules from self molecules. The innate immune system, which plays a pivotal role in the first line of host defense against infection, is equipped with pattern recognition receptors (PRRs) that recognize pathogen-associated molecular patterns (PAMPs) not found in the host and then activate the host's immune response.12 Many PRRs have been identified since toll-like receptors (TLRs) were first identified as PRRs about two decades ago.3 Based on distinct genetic and functional differences, PRRs are currently classified into five families:4 TLRs, nucleotide-binding and oligomerization domain (NOD)-like receptors (NLRs), retinoic acid inducible gene-I (RIG-I)-like receptors (RLRs), C-type lectins (CTLs), and absent-in-melanoma (AIM)-like receptors (ALRs). TLRs and CTLs are located in the plasma membrane, while the NLRs, RLRs, and ALRs are intracellular PRRs.
The recognition by NLRs of PAMPs and damage-associated molecular patterns (DAMPs) from microbial structures or self-or environment-derived molecules leads to the induction of the innate immune response.5 In humans, there are 22 known NLRs,6 all of which are associated with many human diseases.7 Readers who wish to know further basic aspects of NLRs should consult other excellent and recent reviews.47 In this review, we will provide a concise overview of the members of the NLR family and their role in infection, immunity, and disease, especially from clinical perspectives.
CLASSIFICATION AND STRUCTURE OF THE NLR FAMILY
NLR proteins have a common domain organization with a central NOD (NACHT: NAIP, CIITA, HET-E, and TP-2), N-terminal effector domain, and C-terminal leucine-rich repeats (LRRs) (Fig. 1).6 The NACHT domain (consisting of seven distinct conserved motifs, including the ATP/GTPase-specific P-loop, the Mg2+-binding site, and five more-specific motifs) is involved in dNTPase activity and oligomerization.8 The C-terminal LRR domain is involved in ligand binding or activator sensing. The N-terminal domain performs effector functions by interacting with other proteins. There are four recognizable N-terminal domains, which are used to classify NLRs into four subfamilies: the acidic transactivation domain (NLRA), the baculoviral inhibitory repeat-like domain (NLRB), the caspase activation and recruitment domain (CARD; NLRC), and the pyrin domain (NLRP) (Fig. 1).6
The NLRA subfamily includes only one member, the MHC-II transactivator (CIITA). Similarly, the human NLRB subfamily has only one member, NAIP. The NLRC subfamily consists of six members: NLRC1 (NOD1), NLRC2 (NOD2), NLRC3, NLRC4, NLRC5, and NLRX1. NLRC3, NLRC5, and NLRX1 are classified as belonging to the NLRC subfamily due to their homology and phylogenetic relationship, although their N-terminal domains have not been named.469 The NLRP subfamily consists of 14 members, NLRP1-14. No LRR domain is observed in NLRP10, which may indicate a role for this protein as a signaling adaptor rather than as an NLR sensor.7
FUNCTION OF NLRs
The NLRs recognize various ligands from microbial pathogens (peptidoglycan, flagellin, viral RNA, fungal hyphae, etc.), host cells (ATPs, cholesterol crystals, uric acid, etc.), and environmental sources (alum, asbestos, silica, alloy particles, UV radiation, skin irritants, etc.). Most NLRs act as PRRs, recognizing the above ligands and activate inflammatory responses. However, some NLRs may not act as PRRs but instead respond to cytokines such as interferons. The activated NLRs show various functions that can be divided into four broad categories: inflammasome formation, signaling transduction, transcription activation, and autophagy (Fig. 2).4 Below, we describe each function.
Inflammasome formation
Inflammasome is a multimeric protein complex that activates caspase-1.10 Activation of caspase-1 results in the processing and maturation of proinflammatory cytokine interleukin (IL)-1β and IL-18 as well as an inflammatory cell death termed pyroptosis (Fig. 2).10 As IL-1β is a potent mediator of inflammatory responses, its overproduction is associated with many autoinflammatory syndromes, such as gout and periodic fever syndromes, which include Familial Mediterranean fever (FMF) and cryopyrin-associated periodic fever syndromes (CAPS).1112 Pyroptosis is an inflammatory cell death that results in the release of DAMPs and reinforcement of the immune response. Inflammasomes are activated by eight members of NLRs (NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP12, NLRC4, and NAIP) and AIM2, which is not discussed in this review (Table 1).710
Inflammasome formation is triggered by either pathogen-associated or sterile activators. Pathogen-associated activators of inflammasomes include various PAMPs derived from bacteria [pore-forming toxins, lethal toxins, flagellin/rod proteins, muramyl dipeptide (MDP), RNA, and DNA], viruses (RNA and M2 protein), fungus (β-glucans, hyphae, mannan, and zymosan), and protozoa (hemozoin).10 Sterile activators include self-derived DAMPs (ATP, cholesterol crystals, monosodium urate/calcium pyrophosphate dihydrate crystals, glucose, amyloid β, and hyaluronan) and environment-derived stimulants (alum, asbestos, silica, alloy particles, UV radiation, and skin irritants).10 When the eight NLRs detect these PAMPs and DAMPs (Fig. 2), NLRs recruit apoptosis-associated speck-like protein containing a CARD (ASC) via a pyrin-pyrin domain interaction.13 Subsequently, pro-caspase-1 binds to ASC through CARD-CARD domains, which completes the formation of inflammasome.13 NLRP1 contains the CARD domain that can interact directly with procaspase-1 and can thus form inflammasomes without ASC.14 NLRC4, which has no pyrin domain, can form two types of inflammasomes. The recruitment of ASC to the NLRC4 inflammasome results in IL-1β and IL-18 production, while NLRC4 inflammasome formed without the recruitment of ASC results in pyroptosis.15
NAIP and NLRC4 form NAIP-NLRC4 inflammasomes upon recognition of bacterial flagellin and the bacterial type III secretion system.161718 Therefore, NAIP and NLRC4 are linked to susceptibility to bacterial infections.719 NLRP1 inflammasomes are activated by MDP,14 a common peptidoglycan motif in both Gram-positive and Gram-negative bacteria, and by anthrax lethal toxin.20 NLRP3-inflammasome activation is triggered by various PAMPs and DAMPs including alum, silica, ATP, and uric acid.4 NLRP7 can recognize bacterial lipopeptide.21
Signaling transduction
NOD1 recognizes γ-D-glutamyl-meso-diaminopimelic acid (iE-DAP), which is a peptidoglycan component found only in Gram-negative bacteria.22 NOD2 recognizes MDP from both Gram-positive and Gram-negative bacteria.23 Both NOD1 and NOD2 activate the nuclear factor kappa B (NF-kB) signaling pathway, which plays an important role in regulating the host immune response (Fig. 2). Specifically, recognition of cytosolic peptidoglycan ligands allows NOD1/NOD2 to interact with a common downstream adaptor molecule, receptor interacting protein 2 (RIP2), which is a serine/threonine kinase that can activate NF-kB.24 Activated NF-kB can move to the nucleus and enhance transcription of proinflammatory cytokines.2225 In contrast to NOD1/NOD2, NLRC3, and NLRP2/4 act as negative regulators of the NF-kB pathway by modifying tumor necrosis factor (TNF) receptor-associated factor 6 (TRAF6) (Fig. 2).262829 NLRP6 and NLRP12 are also suggested as negative regulators.3031
Transcription activation
Antigen presentation by major histocompatibility complex (MHC) class I and II molecules is central to the function of the adaptive immune system. Therefore, the regulation of these MHC genes and their accessory molecules is crucial for the adaptive immune response. In 1993, NLRA (CIITA) was found to function as a transactivator of MHC class II gene expression. Complementary experiments showed that the NLRA fully corrected the MHC class II regulatory defect of cells from patients with bare lymphocyte syndrome. This rare genetic disorder is characterized by severe combined immunodeficiency and a complete lack of the expression of MHC class II molecules in all tissues.35 Recent studies have shown that NLRC5 plays a crucial role in the expression of the MHC class I gene. NLRC5 induced by interferon-γ acts as a transactivator of the MHC class I gene by assembling regulatory factor X (RFX), cAMP-responsive-element-binding protein 1 (CREB1), activating transcription factor 1 (ATF1), and nuclear transcription factor Y (NFY) on the SXY module in the MHC class I promoter.3637 Although MHC-I and -II expression depends on several transcription factors such as NF-kB, the IFN regulatory factor family, RFX, CREB1, ATF1, and NFY, MHC expression requires the presence of the NLRA and NLRC5.437
Autophagy
Autophagy is a fundamental cellular homeostatic mechanism in which cells autodigest parts of their cytoplasm for removal or turnover. Based on the target of degradation, autophagy has been described as mitophagy, reticulophagy, and pexophagy for autodigestion of the mitochondria, endoplasmic reticulum, and peroxisomes, respectively. In contrast to autophagy, xenophagy refers to an autophagic pathway that targets intracellular bacteria and viruses.38 Autophagy is mediated by unique organelles called autophagosomes, which fuse with lysosomes and allow lysosomal enzymes to degrade the sequestered cytoplasmic materials in autolysosomes.38 Autophagosome formation involves many autophagy-related (ATG) proteins.38
NOD1 and NOD2 can induce autophagy to remove pathogens by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry (Fig. 2).39 NLRX1, located in mitochondria, regulates virus-induced autophagy by interacting with the Tu translation elongation factor of mitochondria (TUFM) that then interacts with ATG5-ATG12 and ATG16L1.40 NLRP4 is known as a negative regulator of autophagic processes through an association with beclin1, one of the key initiators of the autophagic process.41
CLINICAL RELEVANCE OF NLR
In addition to NLRs playing a crucial role in defending against pathogens as a pattern recognition receptor, they also have other functions unrelated to pathogen detection, such as apoptosis and playing a role in early development. Therefore, their abnormalities are linked to various diseases associated with infections, inflammation, and cancer. Understanding their roles in the pathogenesis of certain diseases may help us develop new drugs or new approaches to prevent and treat these diseases. Genome-wide association studies have shown a significant association of polymorphisms of NLR genes with various diseases.710 NLR-associated diseases are summarized in Table 1. Described below are several examples of NLR studies that have shed new insights into disease mechanisms.
Autoinflammatory diseases and NLRs
Autoinflammatory diseases are self-directed inflammatory diseases involving innate immune cells without autoantibodies or autoreactive T cells, whereas autoimmune diseases involve dysfunctional adaptive immune systems producing autoreactive B or T cells.42 Reflecting the important roles of NLRs in inflammation, abnormalities of NLRs have been associated with several (but not all) autoinflammatory diseases. Based on the genes involved, autoinflammatory diseases can be classified into monogenic and polygenic diseases.1142 Monogenic autoinflammatory diseases are caused by abnormalities of a single gene and include FMF (MEFV), CAPS (NLRP3), deficiency of interleukin-1 receptor antagonist (IL1RN), TNF receptor-associated periodic syndrome (TNFRSF1A), hyperimmunoglobulinemia D with periodic fever syndrome (MVK), and Blau syndrome (NOD2).1142 Polygenic autoinflammatory diseases include gout and Crohn's disease.1142 Most autoinflammatory diseases can be explained by the overproduction of IL-1, which occurs as a consequence of inflammasome activation.11 For example, FMF is the most prevalent hereditary autoinflammatory disease in the world and is characterized by recurrent one- to three-day attacks of fever, serositis presenting as abdominal or pleuritic chest pain, and arthritis.1112 FMF is caused by mutations of the MEFV gene, which encodes pyrin.12 Although the association between FMF and pyrin mutations is well established, how pyrin mutation develops into FMF has not been explained. The MEFV gene does not belong to the NLRs family; however, the mutation of the MEFV gene leads to dysregulation of caspase-1, which is similar to the result of NLRP3 (NLR family, "pyrin" domain containing 3) activation.11 Nevertheless, the involvement of NLR genes with some autoinflammatory diseases reflects their important roles in inflammation.
Diseases associated with inflammasome-forming NLRs
Although several NLRs can form inflammasome,710 the NLRP3 inflammasome has been the most studied. Studies found that NLRP3 can recognize a wide range of endogenous and exogenous DAMPs such as uric acid, alum, silica, and asbestos.10 Below are descriptions of how NLRP3 is associated with gout, silicosis, asbestosis, and CAPS and how it is related to the role of alum as a vaccine adjuvant.
Gout is a common metabolic disease described from 2600 BC as podagra, although today it is understood as uric acid arthropathy.43 It is characterized by recurrent, sudden, and severe attacks of pain, redness, and tenderness in joints due to deposition of monosodium urate, a crystallized form of uric acid. However, it was unclear how uric acid could cause inflammatory events until uric acid was found to activate the NLRP3 inflammasome.44 The NLRP3 inflammasome becomes activated in response to uric acid, and its activation induces the formation of IL-1, which leads to the development of gouty arthropathy. Several randomized controlled trials have shown that IL-1-blocking therapy results in significant relief of pain and a reduction in the occurrence of acute flare-ups in gouty patients.11
Silicosis, an occupational disease related to mining and con-struction work, is characterized by pulmonary fibrosis after inhalation exposure to silica. Asbestosis is a similar pulmonary fibrotic disorder following inhalation of asbestos. A recent study showed that silica and asbestos can cause alveolar macrophages to activate NLRP3 inflammasome and produce IL-1β, which leads to the development of pulmonary fibrosis.45
CAPS are rare autoinflammatory diseases, which include familial cold autoinflammatory syndrome, Muckle-Wells syndrome, and neonatal-onset multisystem inflammatory disease.46 Patients with CAPS usually present to physicians with overlapping symptoms such as fever, urticarial skin rash, varying degrees of arthralgia/arthritis, and neutrophil-mediated inflammation.46 CAPS are associated with gain-of-function mutations in the NLRP3 gene,464748 with these mutations inducing an overproduction of IL-1β, which causes CAPS.464748 When IL-1-blocking therapy was given to CAPS patients, significant clinical responses were reported.49
Aluminum hydroxide (alum) has been used as a vaccine adjuvant since the 1920s; however, its mechanism of action was unknown. However, recently, a link has been reported between alum and the NLRP3 inflammasome.50 Alum promotes local necrosis in vaccinated muscle tissue, which leads to the release of DAMPs such as uric acid. DAMPs as well as the alum itself activate NLRP3 inflammasome, which enhances the immune response to vaccine.50 Gaining a better understanding of the role that alum adjuvant plays in the immune response may lead to the development of new vaccine adjuvants.
Single nucleotide polymorphisms (SNPs) in the NLRP3 gene are associated with many disorders such as type 1 diabetes,51 celiac disease,51 psoriasis,52 increased susceptibility to human immunodeficiency virus (HIV)-1 infections,53 and inflammatory bowel diseases.54 NLRP3 is also associated with type 2 diabetes in obese individuals.55 NLRP3 inflammasome in adipose tissue macrophages senses ceramides generated from free fatty acids in obese patients, which can cause obesity-induced inflammation and insulin resistance.55 The development of an effective inhibitor of the NLRP3 inflammasome could, thus, provide a potential therapeutic agent for these NLRP3-associated diseases.56
In addition to the NLRP3 inflammasome, the other NLR inflammasomes are also associated with many diseases. NLRP1 polymorphisms are significantly associated with vitiligo,5758 celiac disease,59 Addison's disease,6061 type 1 diabetes,60 autoimmune thyroid disorders,62 systemic lupus erythematosus,63 systemic sclerosis,64 giant cell arteritis,65 congenital toxoplasmosis,66 rheumatoid arthritis,67 Alzheimer's disease,68 and corneal intraepithelial dyskeratosis.69 NLRP2 gene mutations are associated with Beckwith-Wiedemann syndrome, which is a congenital overgrowth syndrome associated with developmental abnormalities and a predisposition to embryonic tumors.70 NLRP6 expression is predominantly localized in intestinal tissue and is associated with increased susceptibility to colitis and colon cancer in a mouse model.71 Mutations in the maternal gene NLRP7 are associated with recurrent hydatid moles,72 and increased NLRP7 gene expression is observed in testicular seminoma73 and endometrial cancer.74 NLRP12 mutations are associated with atopic dermatitis75 and hereditary periodic fever syndrome.76 NAIP gene mutations are associated with spinal muscular atrophies that are characterized by autosomal recessive disorder and spinal cord motor neuron depletion.77
Diseases associated with non-inflammasome-forming NLRs
Crohn's disease is a chronic inflammatory disease in the gastrointestinal tract. Mutations of the NOD2 gene are associated Crohn's disease, although many patients with Crohn's disease do not have NOD2 mutations.2578 Most NOD2 mutations (93%) in Crohn's disease patients are located in the leucine-rich-repeat region,478 which is responsible for ligand binding. Loss-of-function mutation of NOD2 prevents responses to bacterial MDP in the gut, which might lead to proliferation of commensal or pathogenic gut microbiota in the crypts and disruption of mucosal integrity.4
Mutations and SNPs of the NOD2 gene are also associated with Blau syndrome, which is characterized by familial granulomatous arthritis, uveitis, and skin granulomas.79 Atopic eczema,80 atopic dermatitis,75 and susceptibility to leprosy81 and tuberculosis82 are associated with NOD2 gene mutations. Polymorphisms of the NOD1 gene are linked to asthma83 and inflammatory bowel diseases.84
Bare lymphocyte syndrome type II, also called hereditary MHC class II deficiency, is a severe combined immunodeficiency that results from a lack of expression of MHC class II molecules in all tissues.35 Patients with this disease suffer from multiple infections and frequently die at an early age.35 Loss-of-function due to NLRA mutations leads to reduced expression of MHC class II genes that affect CD4+ T cell function, which in turn causes immune deficiency. Therefore, bare lymphocyte syndrome is an attractive candidate for gene therapy.
Breaks in the NLRA gene were found to occur in B-cell lymphomas, such as primary mediastinal B-cell lymphoma and classical Hodgkin's lymphoma. In addition, NLRA gene alterations have been associated with decreased survival in primary mediastinal B-cell lymphoma.85
CONCLUSION
NLRs are important in the recognition of PAMPs and DAMPs; they also play a crucial role in immune response and pathogen detection. However, NLRs are also important in basic biologic processes, such as apoptosis and embryonic development. Many human NLRs remain poorly characterized and understood. Thus, as we learn more about the function of human NLRs, we will find their pathogenic roles in more diseases and develop novel strategies for treating and/or preventing these diseases.

 , leucine-rich repeat; NOD, nucleotide-binding and oligomerization domain.
, leucine-rich repeat; NOD, nucleotide-binding and oligomerization domain.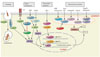

 XML Download
XML Download