

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Limb girdle muscular dystrophy (LGMD) is a clinically and genetically heterogeneous group of disorders characterized by progressive weakness of the pelvic and shoulder girdle muscles. Currently, eight autosomal dominant LGMDs (LGMD1) and 22 autosomal recessive LGMDs (LGMD2) have been identified. LGMD2A is the most prevalent form in many European countries.12 LGMD2A is caused by mutations in the CAPN3 gene (MIM#114240) on chromosome 15q15.1. Calpain-3, encoded by the CAPN3 gene, is a muscle-specific member of the calpain superfamily of calcium-activated neutral proteases. Calpain-3 is associated with regulatory processes such as apoptosis, muscle cell differentiation, sarcomere formation, muscle remodeling, and regulation of the cytoskeleton. However, the pathogenesis of calpainopathy remains a subject of discussion.
The typical clinical presentation of LGMD2A includes progressive proximal muscle weakness, ankle contracture, and winged scapula. However, calpainopathy, caused by CAPN3 mutations, reveals not only LGMD2A phenotypes but also variable clinical phenotypes including pseudometabolic myopathy, eosinophilic myositis, and asymptomatic hyperCKemia.3456 There has been only one small case series of calpainopathy in Korea.7 To identify the characteristics of Korean calpainopathy, we analyzed mutational, clinical, and pathological spectra in Korean patients with CAPN3 mutations.
MATERIALS AND METHODS
Subjects
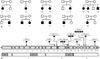
We reviewed our myopathy biobank, which contained 505 muscle and 448 blood samples taken from 896 myopathy patients between March 2004 and August 2011. Firstly, we found four patients from three unrelated families (F1, F2, and F3) who had been previously diagnosed with calpainopathy. F1 and F2 patients were diagnosed with calpainopathy via direct sequencing of CAPN3. Two affected members of the F3 family were confirmed via targeted sequencing for 16 LGMD-related genes (CAPN3, CAV3, DYSF, FKRP, FKTN, LMNA, MYOT, POMGT1, POMT1, POMT2, SGCA, SGCB, SGCD, SGCG, TCAP, and TRIM32). Secondly, we screened the biobank based on the following inclusion criteria: 1) genetically unconfirmed LGMD or hyperCKemia, 2) autosomal recessive or sporadic inheritance, and 3) serum creatine kinase (CK) levels of more than threefold above the upper limit of normal. We selected 51 myopathy patients and performed targeted sequencing of CAPN3. In this way, we identified CAPN3 mutations in nine patients from seven unrelated families (F4, F5, F6, F7, F8, F9, and F10). Then, we analyzed mutational, clinical, and pathological data in 13 calpainopathy patients from ten unrelated families (Fig. 1A). The research protocol was approved by the Institutional Review Board of Gangnam Severance Hospital, Korea. All participants were previously required to sign an informed consent document for mutational analysis.
Targeted sequencing
Genomic DNA from muscle or blood from 51 myopathy patients was extracted with Qiagen DNeasy blood & tissue kits (Qiagen, Valencia, CA, USA). For mutation analysis, the CAPN3 gene was enriched via hybridization capture. The target region included all 24 coding exons and flanking introns of the CAPN3 gene. The captured library was sequenced using an Illumina Hiseq2500 platform with the 2×150 bp paired-end read module. All samples were pooled into a part of a single lane on a single flow cell and sequenced together. A 6-bp index sequence (Illumina, San Diego, CA, USA) and a 6-bp in-house barcode sequence were used to differentiate between samples. We achieved 497.19× mean (or 471.94× median) coverage for all samples. Exonic coverage of the CAPN3 gene at 20× or more was plotted in a standard boxplot. For 99.90% of target regions, capture was at least 20× coverage. Almost 99.98% of regions were captured with at least 10× coverage.
Variant analysis
From HiSeq2500 raw data, sequencing data of all samples were sorted by index and barcode sequences. Sorted fastq files were aligned to the hg19 reference genome with the Burrows-Wheeler Aligner (ver. 0.7.5a) algorithm.8 Output SAM files were converted to BAM files and sorted with SAMtools (ver. 0.1.18).9 Duplicate removal was performed with MarkDuplicates in Picard tools (ver. 1.95). Realignment around known indel sites and base quality score recalibration were performed with the genome analysis toolkit (GATK; ver. 2.6-5) to create the final BAM files.10
Variants were called with the GATK v2.6 Unified Genotyper algorithm.1112 Called variants were filtered based on the following criteria: 1) loci depth ≥10, variant frequency ≥0.5 and 2) loci depth ≥20, variant frequency ≥0.25. Filtered variants were annotated with Annovar (ver. 2013-06-21) using RefGene, dbSNP 135, and the 1000 Genomes Project SNP (2012 April release) of the Asian population and all-population databases. Among called variants, only non-synonymous single nucleotide variants, frameshift indels, and splicing-site variants were chosen. Polymorphisms in Korean populations (n=276) were filtered out. Variants with low functional scores in Polyphen2 (ver. 2.2.2) were considered to be benign mutations and discarded.13 All selected variants were validated by Sanger sequencing.
Clinical assessment
Clinical data included sex; age at symptom onset; family history; initial diagnosis; and presence of winged scapula, lordosis, joint contracture, motor weakness, and muscle wasting. To determine physical disability, we used a Gardner-Medwin and Walton (GMW) scale that had been previously assigned to each patients: grade 0, all normal activities and hyperCKemia; grade 1, normal gait, unable to run freely; grade 2, abnormal gait; grade 3, muscle weakness, climbing stairs with support; grade 4, positive Gower sign; grade 5, unable to rise from floor; grade 6, unable to climb stairs; grade 7, unable to get up from chairs; grade 8, unable to walk independently; grade 9, unable to eat, drink, or sit without support.14 Laboratory findings included serum CK level, nerve conduction study, needle electromyography, electrocardiography, and echocardiography. Serum CK level was expressed as fold over the upper limit of normal values according to the local reference range in 12 patients, excluding F9 patients. Nerve conduction studies and needle electromyography were performed in all 13 patients. Electrocardiography was performed in eight patients (F3a, F3b, F4, F5a, F5b, F5c, F6, and F8). Echocardiograms were performed in F5a and F5b patients.
Pathological assessment
Muscle specimens were obtained for nine patients, taken from the vastus lateralis (F1, F4, F5c, F7, F9, and F10) and the biceps brachii (F5b, F6, and F8). Frozen muscle sections (5-µm thickness) from all muscle specimens were stained with hematoxylin and eosin, modified Gomori trichrome, and reduced nicotinamide adenine dinucleotide-tetrazolium reductase. Protein analysis was performed in seven patients (F4, F5b, F6, F7, F8, F9, and F10). Western blots used antibodies against calpain-3 (Novocastra, NewcastleuponTyne, UK). Muscle specimens were also analyzed by immunohistochemistry using antibodies against the C-terminus of dystrophin (Leica Microsystems, Newcastle upon Tyne, UK), the rod domain of dystrophin (Leica Microsystems), the N-terminus of dystrophin (Leica Micro-systems), dysferlin (Leica Microsystems), α-sarcoglycan (Leica Microsystems), β-sarcoglycan (Leica Microsystems), γ-sarco-glycan (Leica Microsystems), δ-sarcoglycan (Leica Microsystems), α-dystroglycan (Millipore, Billerica, MA, USA), and caveolin (BD Biosciences, San Diego, CA, USA).
RESULTS
Identification of CAPN3 mutations
In the present study, we identified nine different CAPN3 mutations in 13 patients from ten unrelated families. The nine CAPN3 mutations consisted of four missense, two small-insertion, and three splice-site mutations (Table 1), all of which were mainly located in domains III and IV (Fig. 1B). Five mutations were previously reported in the Leiden Database (http://www. dmd.nl/capn3_home.html) or other reports.51516 However, four mutations were not previously reported: c.1524+1G>T, c.1789_1790insA, c.2184+1G>T, and c.2384C>T. Among the 13 patients, two CAPN3 mutations were identified in 11 patients from eight unrelated families (F1, F2, F3, F4, F5, F6, F7, and F8), and only one mutation was found in two patients from different families (F9 and F10). These two patients with a single mutation showed a total loss of calpain-3 protein via western blots (Fig. 2).
Clinical manifestations
The clinical spectra of 13 patients (4 men and 9 women) with CAPN3 mutations were summarized in Table 2. Family history was positive in four families (F3, F5, F7, and F8). The median ages at examination and symptom onset were 33 (interquartile range: 20-37) and 22 (interquartile range: 15-28), respectively. In 12 patients (none from F5), the first symptom was proximal muscle weakness, particularly in the lower extremities. Physical examination revealed joint contracture in nine patients, winged scapula in four, and lordosis in one. Calf pseudohypertrophy was identified in only the F1 patient. The median serum CK levels were 20-fold (interquartile range: 11- to 25-fold) above the upper limit of normal. Nerve conduction studies were normal, and needle electromyography revealed short duration, low-amplitude motor unit potentials, and an early recruitment pattern in all 13 patients. Electrocardiography and echocardiography showed no evidence of cardiacdysfunction in any tested patients.
Not only typical features of LGMD2A but also variable clinical features were found in the Korean patients with CAPN3 mutations. In the F5 family, onset age ranged from 15 to 33 years, even though family members had the same mutations. Additionally, the F5a patient complained of a rapid deterioration in strength during pregnancy. She had four children and recalled a stepwise decline in muscle strength associated with pregnancy. The F7 patient was initially diagnosed with Emery-Dreifuss muscular dystrophy based on early prominent contractures of the elbow and ankle. The F8 patient and his older brother initially visited our hospital for elevated liver enzymes. However, a liver biopsy was normal in his brother, and further laboratory studies showed elevated serum CK level. The patient and his brother did not show clinical signs such as muscle weakness, winged scapula, or ankle contracture. The F9 patient was initially diagnosed with polymyositis based on endomysial inflammatory cell infiltration and relatively abrupt clinical onset.
Pathological assessment
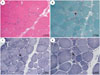
We analyzed muscle biopsies from nine patients in eight families. The muscle specimens of six patients (F4, F5b, F6, F7, F9, and F10) showed nonspecific dystrophic features including varying degrees of fiber-size variability, increased endomysial fibrosis, and degenerative fibers. Among them, four patients (F5b, F7, F9, and F10) had lobulated fibers after staining with nicotinamide adenine dinucleotide tetrazolium reductase (Fig. 3), and the F6 patient had endomysial infiltration of a small number of inflammatory cells. However, three patients (F1, F5c, and F8) showed normal skeletal muscle histology. Muscle specimens were taken from the biceps brachii in F8 and the vastus lateralis in F1 and F5c. When the muscle biopsy was performed, the F8 patient had no clinical symptoms; however, the F1 and F5c patients had mild weakness of the hip and shoulder girdle muscles. Western blot for calpain-3 revealed normal expression in the F4 patient yet nearly total loss in six other patients (Fig. 2). In addition, immunohistochemistry of the muscle specimens showed normal expression of dystrophin, sarcoglycan (α, β, γ, and δ), dysferlin, α-dystroglycan, and caveolin in all 7 patients.
DISCUSSION
We analyzed the mutational, clinical, and pathological spectra of 13 Korean patients with calpainopathy. Among 13 patients, 11 had genetically-confirmed calpainopathy, and two with only a single mutation were diagnosed based on an analysis of calpain-3 protein.
As diagnostic tools for calpainopathy, both protein analysis and genetic tests are complex and require careful interpretation in the light of clinical findings.17 Western blots show normal or almost normal amounts of calpain-3 protein in about 20-30% of patients with CAPN3 mutations.151819 Additionally, protein loss of calpain-3 on western blots is not directly correlated with clinical severity.18 In fact, normal expression of calpain-3 protein was found in the F4 patient with two compound heterozygous mutations. Therefore, mutation analysis of CAPN3 is the gold standard for the diagnosis of calpainopathy. However, previous reports demonstrated that only a single pathogenic mutation is identified in about 10% of calpainopathy patients,21517 as many CAPN3 mutations are located in a deep intronic position, which can be missed during routine gene sequencing.20 In our study, only one mutation was identified in F9 and F10 patients with complete loss of calpain-3 protein.
We studied the clinical and pathological features of Korean patients with calpainopathy. The first clinical symptom usually appeared in the first or second decade of life, and included proximal muscle weakness, winged scapula, and ankle contracture. Half of the patients who received muscle biopsy had lobulated fibers. It is well-known that lobulated fibers increase as the disease progresses, although this is not a specific finding in calpainopathy.16 However, highly variable features were also found in Korean calpainopathy patients. F1, F5b, and F9 patients felt muscle weakness in the third decade. F7, F8, and F9 patients were initially diagnosed with Emery-Dreifuss muscular dystrophy, hyperCKemia, and polymyositis, respectively. Additionally, the F5a patient reported rapid deterioration of muscle and weakness in pregnancy. Although this was the first report on calpainopathy, one retrospective study reported permanent worsening in about half (54%) of 15 LGMD pregnancies.21 However, the cause of exacerbation during pregnancy is not known. Three muscle specimens from patients in this study revealed normal skeletal muscle histology. A previous pathologic report showed normal findings in three preclinical biopsies except in one focal area of necrosis.22 In the present study, although F1 and F5c patients experienced mild weakness of the hip and shoulder girdle muscles, muscle specimens of the vastus lateralis did not have pathologic abnormalities. This result was likely due to the following reasons. Firstly, the predominantly affected muscles in ambulatory patients with calpainopathy are in the posterior compartment of the thigh.23 Secondly, the muscle specimens showed that necrotic and re-generating fibers tended to occur in clusters.16
We did not find a correlation between genotype and phenotype in Korean patients with calpainopathy. It is known that null mutations are usually associated with a more severe phenotype than missense mutations.3152425 In our study, the F7 patient with two null mutations showed severe phenotype, including early clinical onset and prominent joint contracture. However, the F2 patient with two null mutations did not show a more severe phenotype than other patients with missense mutations. In addition, affected members of the F5 family showed clinical variability including onset age, despite the family members having the same genotype.
The present study suggests the possibility of a relatively low incidence of calpainopathy in Korea compared to other countries. To date, our institute, one of the largest myology centers in Korea, has genetically confirmed DYSF mutations, a genetic cause of LGMD2B, in 45 patients (unpublished data). On the other hand, the present study identified CAPN3 mutations in only nine of 51 tested patients with suspicious calpainopathy. However, the relatively small number of patients limited the interpretability of the present study. Therefore, further studies are needed to identify the clinical and mutational findings in a larger cohort.
In conclusion, we identified nine different mutations including four novel CAPN3 mutations in Korean patients. The present study confirmed the high variability of clinical and pathological features in Koreans with calpainopathy.



 XML Download
XML Download