

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Spinal and bulbar muscular atrophy (SBMA, also called Kennedy's disease) is an X-linked and adult-onset motor neuron disease, caused by a CAG repeat expansion with more than 38 repeats in the androgen receptor (AR) gene.1 Affected SBMA patients are characterized by slowly progressive proximal weakness of extremities usually beginning in the shoulder and pelvic girdles, generalized fasciculation and bulbar involvement with onset of neurological symptoms typically around age 30-50 years and may become wheelchair-dependent 20-30 years after onset. Sensory involvement, partial androgen insensitivity and hand tremors have also been reported.2,3,4
Although SBMA is not uncommon in Korea, there is only one study reporting common clinical features and early symptoms with clinical course in 18 Korean patients with SBMA.5 In addition, there are few studies on the rate of disease progression as one of the important disease phenotypes and disease duration as a meaningful parameter in the view of quality of life worldwide.
In this study, we investigated clinical and genetic data of our Korean SBMA patients and compared them with those from various ethnicities. Then, we evaluated the genotype-phenotype correlations between the number of CAG repeats and various clinical characteristics as well as correlations among clinical characteristics in Korean SBMA patients.
MATERIALS AND METHODS
Among 112 patients who were confirmed as having expanded CAG repeats in the AR gene from 1999 to 2013, sufficient clinical data was available in only 40 unrelated male patients. We evaluated the age at onset of symptoms, severity score at onset of symptoms, the interval between onset and diagnosis, disease duration, and the rate of disease progression as clinical measurements. This study was approved by the Institutional Review Board of the Samsung Medical Center.
We assessed nine activities of daily living (ADL) milestones and scored the severity of symptoms according to:6 1=hand tremor (patient's awareness of hand tremor), 2=muscular weakness (initial patient's awareness of muscular weakness in any part of the body), 3=requirement of a handrail (patient is unable to ascend stairs without the use of a handrail), 4=dysarthria (patient is unable to articulate properly and had intelligible speech only with repetition), 5=dysphagia (patient chokes occasionally during meals), 6=use of a cane (patient uses a cane constantly when away from home), 7=use of a wheelchair (patient used a wheelchair when away from home), 8=development of pneumonia (patient develops pneumonia that required in-hospital care), and 9=death. In addition, we modified the ADL milestones by giving symptoms of dysesthesia or muscle cramps a score of 0 since subclinical sensory disturbances contribute to the tremor, which is frequently documented as the initial symptom in SBMA patients.7,8
The age at onset of symptoms was defined as the time of the patient's initial recognition of symptoms, which were included in the ADL milestones from scores 0 to 9. The interval between onset and diagnosis was represented by the time (year) taken from onset of symptoms to genetic diagnosis, and disease duration was defined as interval between onset of symptom and the last follow-up (year). The rate of disease progression was calculated as follows: (the severity score at the last follow-up-the severity score at onset)/(disease duration) for patients who had been followed-up after the diagnosis and (the severity score at diagnosis-the severity at onset)/(the interval between onset and diagnosis) for the other patients (score/year).
The number of CAG repeats in the AR gene was verified by fluorescence polymerase chain reaction (PCR). In brief, genomic DNA was extracted from peripheral blood leukocytes using the Wizard genomic DNA purification kit (Promega, Madison, WI, USA), following the manufacturer's instructions. PCR was performed as described previously9 and the amplicon was analyzed on a capillary electrophoresis-based ABI 3130xl Genetic Analyzer (Applied Biosystems, Foster City, CA, USA). The size of the CAG repeat was cal-culated using GeneScan® analysis software version 2.0.2 (Applied Biosystems).
Genotype-phenotype correlations between clinical measurements and the number of CAG repeats and correlations between clinical measurements were analyzed and determined using Spearman's rank correlation test. Calculations were performed using the statistical software SPSS software version 20 (IBM Software Group, Chicago, IL, USA) and p values of <0.05 were considered statistically significant.
RESULTS
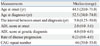
Clinical and genetic data of 40 SBMA patients are summarized in Table 1. The median ages at symptom onset and at diagnosis were 44.5 (20.0-71.0) and 52.5 (34.0-74.0) years, respectively, and median interval between onset and diagnosis was 5.0 (0.25-50.0) years. Median number of CAG repeats was 44 (39.0-55.0). ADL score at onset was 2.00 in most patients, but the most common score at diagnosis was 6.00 (Fig. 1). Of 40 patients, only 14 patients were followed up after diagnosis and their median disease duration and median period from diagnosis to the last follow-up was 11.5 (2.0-51.0) and 5.0 (1.0-11.0) years, respectively. Among 14 patients followed up after genetic diagnosis, one patient had been expired due to recurrent pneumonia. Median rate of disease progression for a total 40 patients was 0.23 (0.0-4.0) score/year.
In correlation analysis between genotype and phenotype, the number of CAG repeats showed significant inverse correlation with the age at onset of symptoms (r=-0.407, p=0.009) (Fig. 2A). In addition, the number of CAG repeats was also inversely correlated with the age at onset of muscle weakness (n=25, r=-0.428, p=0.033), but not with other parameters including the score of severity at onset (r=0.174, p=0.283) and the rate of disease progression (r=0.006, p=0.971).
In the correlation analysis between clinical characteristics, patients with an earlier onset of symptoms had a marginal trend with slow disease progression (r=0.367, p=0.020) (Fig. 2B). However, no significant correlation was observed between the age and the score of severity at onset (r=0.007, p=0.966) as well as the score of severity at onset and the rate of disease progression (r=-0.129, p=0.427).
DISCUSSION
There have been a study for Korean patients with SBMA and several reports from various ethnicities, however, this is a study of the largest Korean cohort of 40 patients with SBMA reported to date, as shown in Table 2. The major symptoms of SBMA are weakness or atrophy,10 but is often preceded by nonspecific symptoms such as postural tremor and muscle cramps.11 In the current study, dysesthesia or muscle cramps of ADL score 0 and hand tremor of ADL score 1 as initial symptoms were not uncommon with the proportion of 30% (12/40), although muscle weakness as initial symptoms was most common with 62.5% (25/40). All muscle cramps were noticeable in the face and all dysesthesia were affected distally in the legs as known so far and these symptoms are the main clinical features distinguishing SBMA from other motor neuron disease, although genetic diagnosis is indispensable for diagnosis.11 Typically, affected individuals require a wheelchair 15-20 years after onset of weakness. According to our scoring method for severity of symptoms, the rate of disease progression of these typical progression aspect could be calculated to 0.25-0.33 score/year (ADL score 7-ADL score 2/15-20 years), and the current study revealed similar or slightly slower rate of disease progression of 0.23 score/year.
When comparing this Korean study with the previous Korean study by Lee, et al.,5 the present study showed that the age at diagnosis was similar but the number of CAG repeats and disease duration were shorter. The shortened disease duration in the current study may be caused by statistical differences between the two studies, such as the number of cases or the expression method (mean and median). However, the possibility that the diagnosis was made earlier in this study due to the improvements of clinical recognition and advances of diagnostic technologies in SBMA cannot be excluded. The shortened number of CAG repeats in the current study is likely caused by the geographical difference between the Busan province, where patients were mainly recruited in the study by Lee, et al.,5 and Seoul, where patients were mainly recruited in the current study or difference in the number of cases.
Previous studies with more than 30 SBMA cases showed a significant inverse correlation between the number of CAG repeats and the age at onset of symptoms, whereas other previous studies with a smaller number of cases including a study for Korean patients, did not show this correlation.3,5,6,8,12,13 In the present study, we recruited 40 patients and the number of CAG repeats showed a statistically significant inverse correlation with the age at onset of symptoms as well as muscle weakness. In contrast, the number of CAG repeats was not correlated with either the disease duration or the rate of disease progression.
There have been a few reports regarding the genotype-phenotype correlation in patients with SBMA,2,3,5,6,8,10,12,13,14,15 while few studies have evaluated correlations between clinical characteristics. Therefore, we analyzed the correlations between clinical characteristics, including the age at onset of symptoms, severity at onset of symptoms and rate of disease progression. As demonstrated in the results, the age at onset of symptoms was marginally correlated with the rate of disease progresses. To our best knowledge, this is the finding which has not been evaluated or reported previously. The reason for this finding is most likely due to the fact that the older the patient with SBMA is, the faster the aging process accompanied with structural and functional changes of muscle with deteriorated muscle weakness proceeds.
Although patients with a younger onset have slower rate of disease progression, ages 30 through 50 is usually the most productive period and involves family support. Additionally, suffering from SBMA at this period not only affects an individual's quality of life, but also leads to considerable socioeconomic loss by the expenditures of health insurance and the loss of productivity of patients and their caregivers. Furthermore, a slow and gradual course of SBMA with insignificant initial symptoms makes diagnosis more difficult, leading to the person being undiagnosed after a long time and period from onset of symptoms often ranging up to several decades as shown in previous studies as well as this study.3,5,6,14,15 Considering these findings, an early diagnosis of SBMA through genetic screening is needed, and ongoing development of new therapeutic strategies could make genetic screening more justified.16
In conclusion, this study reaffirms the inverse correlation between the age at disease onset and the number of CAG repeats. Although the number of cases is small compared to previous studies on other ethnicities, this report would be the largest and recent SBMA cohort in Korea, and the number of CAG repeats and interval between onset and diagnosis in our study might most well represent Korean patients with SBMA. In addition to, it is of note that the earlier onset of disease symptoms is correlated with the slower disease progression in spite of limitation of data based on the patients' subjective memory. Furthermore, large-scale cohort studies with various ethnicities are needed.



 XML Download
XML Download