

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophilia A is caused by a lack of factor VIII (FVIII) in males due to X-linked recessive genes, and comprises 80-85% of all hemophiliacs.1 FVIII concentrates are essential for treating patients with hemophilia A and have evolved from plasma-derived FVIII concentrates to recombinant FVIII (rFVIII) concentrates in the last two decades. The production of FVIII using recombinant DNA technology has emerged as an alternative to plasma-derived FVIII concentrates, which introduce problems of limited yield and the possibility of propagating various viral diseases. The rFVIII concentrates first used clinically included Kogenate® (Bayer Healthcare, Berlin, Germany), Kogenate-FS® (Bayer Healthcare, Berlin, Germany), and Recombinate® (Baxter Healthcare Corporation, Westlake Village, CA, USA).2,3,4 Octocog alfa, moroctocog alfa, and turoctocog alfa, which are rFVIII concentrates developed using plasma/albumin-free method, were introduced consecutively as third generation treatments.5,6,7 Despite the recent development of several rFVIII concentrates, their use is still limited in some countries, where clinicians face difficulties in treating patients with hemophilia A.
Beroctocog alfa is a second generation rFVIII manufactured by removing the B-domain from FVIII and is the second commercially available B-domain-deleted (BDD) rFVIII concentrate after ReFacto® (Wyeth Pharmaceuticals, Collegeville, PA, USA). This prospective clinical trial was conducted to evaluate the efficacy of beroctocog alfa in treating acute hemorrhage and for prophylaxis during surgical procedures in patients previously treated for hemophilia A. This trial also evaluated the pharmacokinetic (PK) characteristics and safety aspects thereof in relation to FVIII inhibitor production, immunological reactions, viral dissemination, and the occurrence of adverse events (AE).
MATERIALS AND METHODS
Subjects
The eligibility criteria comprised an age of ≥12 years, diagnosis of severe hemophilia A with a baseline FVIII level ≤2% of normal (≤1% for the PK test), and prior treatment with FVIII concentrates for 150 exposure days or greater. Exclusion criteria included the presence of an FVIII inhibitor of 0.6 Bethesda units (BU) or higher; presence of antibodies to rFVIII, mouse IgG, or Chinese hamster ovary (CHO) cells; a history of hematological disorders or blood coagulation diseases other than hemophilia A; presence of human immunodeficiency virus (HIV) antibody; CD4 lymphocyte cell count <400/µL; or a history of hypersensitivity to FVIII concentrates.
This trial was approved by the Korea Food and Drug Administration
and the Institutional Review Boards of the participating
institutions. Written informed consent was obtained prior to the study from the patients or an acceptable legally representative. This trial was conducted in accordance with the Declaration of Helsinki8 and was registered at www.clinicaltrials.gov (NCT01568580).
Study drug
Beroctocog alfa, developed by the Green Cross Corporation (Yongin, Korea), is manufactured by removing the B-domain from FVIII and introducing the genetic sequence of this FVIII to CHO cells to establish a recombinant cell line whose cells are massively cultured through the suspension method using serum free medium without human or animal proteins to produce massive amounts of FVIII.9 FVIII generated in this way is separated for purification and finished as a product in a lyophilized formulation. Beroctocog alfa is unique from other second generation rFVIII products in that human albumin is in the final formulation but the cell culture process is free of albumin or other human or animal source proteins. The purification process is comprised of viral removal processes with an affinity column and three consecutive ion exchange columns. In the final lyophilization manufacturing process, a human serum albumin excipient, which is validated for absence of pathogens and virals, is included in the lyophilized formulation. The molecular weight of beroctocog alfa is approximately 170 kDa and includes FVIII consisting of 1425 amino acids. The study drug was provided as a sterile, freeze-dried powder of 500 IU per vial.
Study design
This prospective, open-label, non-comparative, multicenter trial was conducted to evaluate the efficacy and safety of rFVIII, beroctocog alfa, in previously treated patients with hemophilia A. This trial enrolled 71 patients at four sites in Korea and was completed in 21 months between December 2004 and September 2006.
This trial consisted of three parts: an acute hemorrhage treatment study (study 1), prophylaxis for surgery study (study 2), and a PK study (study 3). All subjects eligible for study 1 were administered 10-50 IU/kg of beroctocog alfa intravenously whenever bleeding occurred at home. The administered dose was determined by consulting past doses in each subject and the guidelines on FVIII doses set by the World Federation of Hemophilia.10 The study drug was administered by patients as soon as possible when there was a hemorrhagic symptom, and an additional dose was taken if hemostatic effects were insufficient. If the additional dose still had insufficient hemostatic effects, the subject visited the institution to assess FVIII inhibitor formation. The last visit was planned when 100 exposure days to the study drug were achieved or when the study drug administration period reached one year. When a subject participating in study 1 was hospitalized for treatment of major bleeding during the study period, the dose and administration period were determined based on the investigator's judgment.
For study 2, subjects who were enrolled in study 1 and needed to receive a surgical procedure during the study period were administered the study drug prior to the surgical procedure to prevent hemorrhage. The dose and administration period were determined at the level capable of preventing hemorrhage according to the type of procedure and condition of the subject based on the judgment of the investigator. A surgical procedure was defined as any treatment involving not only major surgery but also relatively simple minor surgery, such as tooth extraction, in the oral surgery department, the orthopedics department, or the general surgery department. The investigator evaluated hemostatic effects, as well as the amount of hemorrhage during and after the procedure, the volume of transfusion, and any AEs. When the subject recovered after the procedure, study 1 was continued until the end of the scheduled study period.
Study 3 proceeded with subjects who were enrolled in study 1 and agreed to participate in the PK study. After at least a 72-h washout period, 50 IU/kg of the study drug was administered over 15 minutes by intravenous infusion at the study institution at baseline and 6 months after administration. Blood samples were collected 15 times before starting drug administration until 48 hours thereafter (at a regular time interval) to measure FVIII: C levels. Adverse event evaluation and clinical tests were performed until 48 hours after administration. Subjects who entered the PK study at baseline and participated in study 1, continued with home treatment and re-performed the PK study after 6 months.
Clinical assessments
The primary endpoint of studies 1 and 2 was the investigator's final evaluation of hemostatic effects. In study 1, the subjects recorded the time and date of hemorrhage, the site of hemorrhage, the time and date of administration and dose, and hemostatic effects on a 4-point scale of excellent (totally hemostatic), good (almost hemostatic), moderate (not enough but hemostatic effect recognizable), and none (hemostatic effect not recognizable) when administering the study drug at home, 24 hours after the administration, or right before additional administration in a diary. The subjects visited investigators every month to evaluate hemostatic effects of the home on-demand treatment. Then, the investigators prepared a monthly review of the subject's daily record and evaluated the hemostatic effects on a 4-point scale based on a physical examination and other tests. At the end of the study, a final evaluation of the hemostatic effects was prepared based on the monthly evaluations by the investigators. Efficacy rate was defined as [(number of subjects who responded "excellent" or "good")/(total number of subjects)]×100.
The secondary endpoints of study 1 were the dose frequency, the dose of the study drug used monthly and the dose used to treat a single hemorrhage, the subjects' evaluation of hemostatic effects and its recovery rate, the formation of FVIII inhibitor, immunological reactions (anti-rFVIII antibody, anti-mouse IgG antibody, anti-CHO antibody, CD4, CD8, and CD4/CD8), viral infection positivity [HIV-antibody, anti-HAV (IgG, IgM), HBsAg/anti-HBs, and HCV-antibody], and AEs. Blood samples were drawn every 3 months, including at baseline, to assess development of an FVIII inhibitor, recovery rate, complete blood count, clinical chemistry tests, immunological reactions, and viral infections, and were analyzed at a central laboratory (Green Cross Medical Foundation, Yongin, Korea). The activity of plasma FVIII was assessed by a one-stage clotting assay at a central laboratory. FVIII inhibitor level was initially measured using the Bethesda method. If the FVIII inhibitor level was >0.6 BU according to the results of Bethesda assay, it was retested using the Nijmegen assay.11 If the retest results showed >0.6 BU of the FVIII inhibitor, the FVIII inhibitor was considered to have formed.
Statistical analysis
The recruiting goal of this trial was to include more than 50 previously treated patients as recommended in the European Medicines Agency's guideline. The subjects were classified into four populations: the intention-to-treat (ITT) population, all subjects who were treated according to the study protocols; a per-protocol 1 (PP1) population, who satisfied 100 days of exposure to the study drug or were administered the study drug for an entire year; the per-protocol 2 (PP2) population, who did not commit any serious protocol violation and completed the clinical trial according to the protocol; and the safety population, who had at least one administration of the study drug. The analyses of hemostatic efficacy were conducted for the ITT population in principle, and additional analyses were performed on the PP1 and PP2 populations. The analyses of baseline characteristics of the subjects and laboratory tests targeted the ITT population, while analyses of AEs targeted the safety population (Fig. 1).
Baseline characteristics, hemostatic efficacy, and safety variables are presented using descriptive statistics. The PK parameters between each visit were compared using the Wilcoxon-signed rank test to confirm statistical differences. All p-values<0.05 were considered significant.
RESULTS
Subjects
A total of 91 subjects with severe hemophilia A were screened for participation in this study, and 20 were excluded; thus, 71 patients were enrolled (Fig. 1). Of these, 14 subjects dropped out, and 57 subjects completed the acute hemorrhage treatment study (study 1). Of the 71 subjects enrolled in study 1, 10 entered the prophylaxis for surgery study (study 2), and 12 entered the PK study (study 3).
The ITT population included 70 subjects who had evaluable hemostatic efficacy data. All 70 subjects were Asian males. Their mean age was 31.9±9.6 years, and three of the 70 patients (4.2%) were adolescents (12-18 years of age). The median duration of previous exposure to FVIII was 18.9±5.9 years. A total of 52/70 subjects (74.2%) were positive for hepatitis C.
Hemostatic efficacy of acute hemorrhage treatment
During study 1, a total of 5142 acute hemorrhages occurred in all subjects. Bleeding episodes were most frequent in joints (78.1%), followed by muscles (11.5%), multiple sites (3.7%), oral (3.6%), soft tissue (1.6%), and others (1.3%). The majority of acute hemorrhages were mild or moderate, and there were 2 episodes of major bleeding (traumatic intracerebral hemorrhage, upper gastrointestinal bleeding).
The investigators' final evaluations of hemostatic effect for acute hemorrhage were excellent in 35 subjects (50%), good in 26 subjects (37.1%), moderate in 9 subjects (12.9%), and none in 0 subjects (0.0%). The overall efficacy rate was 87.1% [95% confidence interval (CI) 79.3-95.0%]. The investigators' evaluations of hemostatic effect for major bleeding were excellent in 1 case and good in 1 case, showing 100% efficacy rate (Table 1). There were a total of 6179 cases in which hemostatic effects were evaluated: the efficacy rate was 72.3% (95% CI 71.2-73.5%), with excellent efficacy in 1425 cases (23.1%), good efficacy in 3045 cases (49.3%), moderate efficacy in 1583 cases (25.6%), and no efficacy in 126 cases (2.0%).
The majority of acute hemorrhages were treated with one (86.2%) or two (10.0%) administrations of the study drug. In terms of site of hemorrhage, bleeding was stopped by administering the study drug once in 87.1% of cases involving joints, 84.8% involving muscles, 82.3% involving multiple sites, 82.8% involving oral sites, 78.3% involving soft tissues, and 70.6% involving other sites (Table 2). The subjects were administered the study drug 88.30±31.92 times on average, and the mean dose administered per single dose was 28.55±6.53 IU/kg. An average of 1.20 times of 34.31 IU/kg was administered to treat a single hemorrhage. The subjects were administered the study drug 9.18±3.67 times (17710±9325 IU) every month.
The baseline recovery was 2.16±0.85%/(IU/kg), ranging from the minimum of 0.58%/(IU/kg) to the maximum of 3.98%/(IU/kg). The recovery at the close-out visit was 1.70±0.77%/(IU/kg), which when compared to the recovery at the baseline, showed statistically significant decrease (p=0.0015). However, considering the fact that the normal range of FVIII: C is 60-150%, the difference of FVIII: C being 60-150% before and after administering 50 IU/kg of FVIII concentrate, as in this clinical trial, makes the recovery of 1.2-3%/(IU/kg) which could lead to the interpretation that the recovery change of the study drug represents a change within a normal range.
Hemostatic efficacy of prophylaxis for surgery
Ten subjects received 12 surgical procedures in this trial. The surgical procedures included arthroplasty (two cases), arthrolysis (one case), hemorrhoid operation (two cases), tooth extraction (three cases), tooth extraction site suture (one case), subgingival curettage (one case), epidermal cyst incision and drainage (one case), and hordeolum incision and drainage (one case). The median dose administered per surgical procedure was 8750 (2000-189000) IU, and hemostatic efficacy by the investigators for the surgical procedures was excellent in seven cases (58.3%) and good in five cases (41.7%), showing a 100% efficacy rate.
Safety
A total of 88 subjects who were administered the study drug more than once were included in the safety analysis. Three subjects who had a surgical procedure after the clinical trial close-out and received new subject numbers through re-screening were analyzed as new subjects. The subjects acquired 6397 rFVIII exposure days during studies 1 and 2, and 82.5% of the subjects had more than 50 rFVIII exposure days.
In this trial, 52 of 88 subjects (59.0%) experienced 168 AEs. Eighteen serious AEs (10.7% of AEs) occurred in 11 subjects who all recovered. Five subjects dropped out because of AEs. Among the 18 serious AEs, two (mild dyspnea and facial edema) in one subject were related to the study drug. There were 43 AEs (25.6%) that were related to the study drug and were evaluated as definite/possible/probable. Of the 127 AEs that occurred while treating acute hemorrhage, 26 (20.5%) in 16 subjects were related to the study drug: anti FVIII antibody positivity (six events), urticaria (three events), palpitations (two events), nausea (two events), and facial edema (two events), anemia (two events), and others (one event each). Of 24 AEs that occurred during prophylaxis for surgery, seven (29.2%) in two subjects were related to the study drug: infection site swelling (three events), phlebitis (two events), and others (one event each). Ten of 17 AEs (58.8%) in six subjects during the PK study were related to the study drug, such as headache, nasal congestion, sputum retention, and throat irritation (two events for each), as well as nausea, axillary pain, blood IgG increase, abnormal liver function test, dizziness, somnolence, pyuria, cough, and sneezing (one event for each). The FVIII inhibitor was negative before study drug administration (<0.6 BU) in the entire ITT population, and only one subject formed de novo FVIII inhibitor for an occurrence rate of 1.4%; the one-sided 95% upper confidence limit was 3.85%.
Among immunological reactions, 11 (18.6%) of 59 subjects turned from normal to abnormal for CD4; this transition also took place for CD8 in three (4.9%) of 61 and for CD4/CD8 in five (9.1%) of 55. These reactions did not show statistical significance before or after administration of the study drug. After study drug administration, we performed enzyme linked immunosorbent assay (ELISA) assay, in which anti-rFVIII Ab in four (5.7%) of 70 subjects and anti-mouse IgG in one 1 (1.4%) of 70 subjects turned from negative to positive.
Anti-HAV IgG turned from negative to positive in four of 10 subjects (40%), although anti-HAV IgM was negative in these four subjects. Anti-HBs turned from negative to positive in two of 17 subjects (11.8%). A positive conversion rate for HIV Ab, anti-HAV IgM, HBs Ag, and HCV Ab was not observed in any subject.
Pharmacokinetics
The results of the PK parameters are presented in Table 3. No differences were observed between visits as the final elimination half-life was 13.3 h and 12.6 h at baseline and 6 months, respectively. The maximum activity in blood (Cmax) showed a mean of 176.7% at baseline but a mean of 137.6% at 6 months, and the blood activity-time area under curve (AUC48h) showed a mean of 1971.9 h·% at baseline but decreased to 1356.0 h·% at 6 months (p=0.003 and p=0.013, respectively). Recovery decreased from a mean of 137.0% at baseline to a mean of 112.2% at 6 months (p=0.026).
DISCUSSION
Beroctocog alfa (GreenGene®, Green Cross Corporation, Yongin, Korea) is a newly developed second generation rFVIII manufactured by removing the B-domain from FVIII, and is the second commercially available BDD rFVIII concentrate after ReFacto®. In this clinical trial, beroctocog alfa showed excellent efficacy in the treatment of acute hemorrhage and as prophylaxis for surgical procedures in patients previously treated for severe hemophilia A. Beroctocog alfa also revealed an acceptable safety, based on its AE profile.
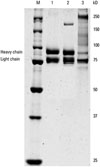
Previous results of phase I and II studies to evaluate the PK parameters and safety profiles of 25 and 50 IU/kg of beroctocog alfa through single and repetitive administrations in 12 hemophilia A patients demonstrated comparable safety and PK profiles in comparison to those of commercial plasma derived FVIII (active control).12 No serious AEs were observed, and the PK parameters showed dose-independent effects in the 25 and 50 IU/kg dose ranges. Interestingly, beroctocog alfa is a highly homogenous rFVIII product with a heavy chain (A1 and A2 domain) and a light chain (A3, C1, and C2 domain) at 90 and 80 kDa.13 Paik, et al. previously reported beroctocog alfa to be homogenously secreted and highly purified FVIII. Sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) results demonstrated beroctocog alfa has no heterogeneous B domain, such as full length FVIII, nor a single-chain protein, as in other commercial rFVIII concentrates (Fig. 2). By separating itself from other FVIIII products that are observed in the SDS-PAGE to be heterogeneous in many form of FVIIII, it acquires a distinctive homogenous characteristic.14,15,16 The homogeneity of the study drug may contribute to lessening concerns for safety and quality control of rFVIII products.16 Accordingly, it is not because beroctocog alfa does not utilize albumin during the cell culture process, making this product different from ReFacto®, that it becomes highly attractive; instead, its homogeneity sets it apart from other rFVIII products.
The hemostatic efficacy rate of beroctocog alfa was 87.1% in the present study, and that of Kogenate FS® was reported to be 82.9%.17 The efficacy rates of ReFacto® and octocog alfa were 86%5 and 86%,6 respectively. In addition, the rate of achieving hemostasis for a single hemorrhage with two or fewer administrations of the study drug was relatively higher than that of other rFVIII concentrates. Instances of administering rFVIII concentrate only once or twice to treat a single hemorrhage was 96.2% for the study drug, 88.7% for Kogenate FS®, 91.2% for Recombinate®,18 86.7% for ReFacto®,5 92.5% for Xyntha® (Pfizar Inc., New York, NY, USA),5 and 93.0% for octocog alfa.6 Although a direct comparison of the efficacy rate between the study drug and other rFVIII concentrates could not be performed, the study drug had comparable hemostatic efficacy with previous rFVIII concentrates. Additionally, the hemostatic effects were excellent in 12 surgical procedure cases, verifying the efficacy of the study drug for inhibiting and preventing hemorrhage symptoms.
Of the 127 AEs that occurred during acute hemorrhage treatment, 26 were related to the study drug, and out of the 24 AEs that occurred during surgical procedures, seven were related to the study drug. The common AEs were anti FVIII antibody positivity (6 events), urticaria (3 events), and infectious site swelling (3 events). The clinical trial results of ReFacto® showed that 70 AEs related to the study drug occurred in 31 of 113 subjects (27.4%), and they mostly consisted of nausea (11 events), dyspnea (7 events), headache (5 events), vasodilation (5 events), asthma (3 events), angina pectoris (2 events), and anorexia (2 events).19 Although beroctocog alfa showed a higher incidence of AEs than ReFacto®, only two serious AEs (mild dyspnea and facial edema) in 1 subject were related to the study drug, and most of AEs were not serious and manageable. No significant changes in immunological reactions or viral propagation were observed. Therefore, beroctocog alfa appeared to be as safe as other rFVIII products for use.
The major PK parameters of administration of the study drug after 6 months and the results of previous research are similar. The parameters for the first administration period showed differences from those at 6 months and the results of previous research.12 AUC48h and recovery between the first administration period and administration after 6 months was 0.78, 0.70, and 0.85, in terms of the ratio of the value at 6 months to baseline for Cmax, AUC48h, and recovery. The exact cause of the change in the PK parameters between baseline and 6 months was unclear. Antibody formation, weight change, change in the test instrument or test method, or change in the manufactured lot of the study drug could be suspected reasons for the differences. Almost no change in weight and age was observed. There was also no change in test method and the manufacturing process. The decrease in recovery with time when administering the same rFVIII concentrate to the same patient is caused by antibody formation.2 However, the results of conducting previously identified antibody tests showed no antibody formation in the subjects that participated in this clinical trial.20 Considering that the degree of recovery of various recombinant concentrates in published reports varied from 73.5% to 135.0%17,20 and that the recovery of the same concentrates in existing studies (mean of 113.0%)12 was similar to the recovery at 6 months, we suggest that the possibility of over-estimating recovery at baseline may account for the discrepancy. When administering blood-derived FVIII concentrates or rFVIII concentrates, the dose is determined to achieve FVIII activity in blood of 100% of the normal value. As this is calculated by assuming 100% recovery, the clinical significance can be considered the same if recovery is 100% or more.21 When beroctocog alfa was administered at baseline and 6 months, the mean recovery was 100% and the administration of 50 IU/kg beroctocog alfa for 6 months reached a recovery rate of 100% or more.
Previous studies have demonstrated that the B-domain of FVIII is dispensable for in vitro procoagulant activity.22 However, there are several concerns about BDD-rFVIII concentrates. A meta-analysis reported a shorter half-life of BDD-rFVIII compared with full length rFVIII concentrates.23 This meta-analysis had some limitations in that the analysis included non-randomized observational studies in a single cohort. In contrast, two randomized controlled PK studies failed to confirm a difference in PK parameters between the BDD-rFVIII concentrate ReFacto® and full-length FVIII and showed a bioequivalence of BDD-rFVIII concentrates and full-length FVIII.24,25 Furthermore, the incidences of de novo FVIII inhibitor for Xyntha®, ReFacto®, and beroctocog alfa are only 1.5% (three of 187 subjects for both Xyntha® and ReFacto®)5 and 1.4% (one of 70 subjects for beroctocog alfa), and these incidence rates of inhibition by BDD-rFVIII concentrates are comparable with full length FVIII concentrates.
There are some limitations to this study. We reported the efficacy and the safety of beroctocog alfa of on-demand treatment instead of prophylactic treatment because only the former was permitted as a clinical trial under Korean regulations in 2004. Also, the high rate of drop out subjects made this study less of a well-controlled trial. Only 36 of the 71 (50.7%) enrolled subjects completed the study according to the protocol, although hemostatic efficacy was consistently high in the ITT, PP1, and PP2 populations. Additionally, this study was conducted several years ago and the publication was delayed due to several reasons; however, little has changed in the management of hemophilia A.
In conclusion, this clinical trial demonstrated that beroctocog alfa is safe and effective in treatment of acute hemorrhage and as a surgical prophylaxis in patients with hemophilia A. Beroctocog alfa provides another option for treating adolescents and adult patients with hemophilia A.




 XML Download
XML Download