

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hypoparathyroidism, deafness and renal dysplasia (HDR, OMIM 131320) syndrome is a rare condition inherited as autosomal dominant trait.1 Clinically, hypoparathyroidism is characterized by symptomatic or asymptomatic hypocalcemia with undetectable or normal serum concentrations of parathyroid hormone (PTH).2 The sensorineural hearing loss is bilateral and most pronounced at higher frequencies, and hearing loss is typically moderate to severe and present at birth.3 Renal anomalies include developmental anomalies, such as, renal dysplasia, hypoplasia, aplasia, cystic kidneys, vesicoureteral reflux (VUR), and functional anomalies.4 The disorder may be genetically heterogeneous, and the correlation between HDR associated GATA3 mutations and phenotypes has not been established.5,6 Here, we report on a symptomatic 32-day-old Korean male with HDR syndrome in whom a novel mutation of the GATA3 gene was identified.
CASE REPORT
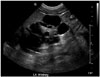
A 32-day-old male was admitted to our hospital for the evaluation of repeated seizures over the previous 3 days. The patient was born after 40 weeks of gestation by vaginal delivery after an uneventful pregnancy and weighed 2.93 kg. He was the first-born baby of healthy Korean parents, and had no dysmorphic features. However, renal malformation was suspected based on fetal ultrasound imaging findings obtained at 24 weeks. A physical and neurological examination at admission revealed no abnormal findings. Laboratory testing showed the following results: plasma calcium 4.6 [reference range (RR), 8.8-10.8] mg/dL; ionized-calcium 0.53 (RR, 0.95-1.5) mmol/L; magnesium 1.8 (RR, 1.6-2.6) mg/dL; phosphorous 9.8 (RR, 3.8-6.5) mg/dL; alkaline phosphatase 1511 (RR, 60-360) IU/L; intact-PTH 5.59 (RR, 9-65) pg/mL; 25-hydroxy vitamin D 11.96 (RR, 8.0-51.9) ng/mL; and 1,25(OH)2 vitamin D3 7.44 (RR, 19.6-54.3) pg/mL. A multicystic left dysplastic kidney and VUR were detected by ultrasound after birth (Fig. 1). Brain magnetic resonance imaging and electroencephalography revealed normal findings. Auditory brainstem response (ABR) testing revealed that the patient had moderate sensorineural deafness, with hearing losses of 80 dB at the mid and higher frequencies for both ears. Echocardiography and electrocardiography findings were abnormal, including small secundum atrial septal deftect and first degree atrioventricular block. Based on the biochemical results obtained and clinical findings, a presumptive diagnosis of HDR syndrome was made. Written consent was obtained from the parents for the blood samples. Direct sequencing identified a heterozygous deletion, c.153del in exon 1 of the GATA3 gene (Fig. 2). This deletion is predicted to cause a frameshift that results in a premature stop codon (p.Phe51Leufs*144). The c.153del mutation is novel and has not been previously described in HDR syndrome. Furthermore, the c.153del mutation was not detected in either parent.
After confirming hypoparathyroidism, calcitriol and calcium therapy was started, and the therapy successfully resulted in normal serum calcium and phosphate values. Subsequently, he remained seizure free without antiepileptic medication. At his 21-month follow-up (aged 22 months), he had normal developmental milestones and growth parameters.
DISCUSSION
The described case was diagnosed as HDR syndrome based on the patient's clinical manifestations and molecular analysis. The triad of HDR syndrome is variably manifested by patients with GATA3 abnormalities, and is caused by haploinsufficiency of the GATA3 gene located on chromosome 10p15 due to a heterozygous inactivating mutation in the GATA3 gene.7,8 Human GATA3 expression has been detected in developing parathyroid glands, inner ears, kidneys, thymus, and central nervous system.9,10 Ali, et al.11 classified GATA3 mutations into three classes based upon their functional consequences on DNA binding. The first class of mutations results in loss of DNA binding due to loss of the carboxyl-terminal zinc finger and represents more than 90% of all mutations. The second class of mutations reduces DNA-binding affinity, and the third class does not alter DNA binding or affinity but probably leads to conformational changes.11 In our case, the deletion mutation (c.153del) disrupted the N-terminal coding region of GATA3, and possibly, and adversely affected stability of the GATA3 DNA binding and interactions between GATA3 and other zinc finger proteins.
Over 90% of patients with HDR syndrome present hypoparathyroidism and sensorineural deafness, and more than 80% show urinary tract and renal abnormalities.6 Our patient had the typical triad of HDR syndrome. He showed symptomatic hypocalcemia with a low serum PTH concentration, which resulted in a diagnosis of hypoparathyroidism. Bilateral sensorineural hearing loss requiring cochlear implantation was diagnosed at one month of age and renal abnormality including VUR was detected after birth. However, the diagnosis of HDR syndrome is sometimes difficult, because patients with the same genotype and laboratory findings of symptomatic HDR syndrome have been found to be asymptomatic.7 Therefore, we suggest that HDR syndrome should be considered in the differential diagnosis in patients with hypoparathyroidism, and that renal ultrasound or ABR testing be performed to prevent a missed diagnosis. In addition to HDR triad, it is noteworthy that our patient showed heart defect, because congenital cardiac abnormality has rarely been reported in HDR syndrome. This abnormality in the patient would be irrelevant to the GATA3 haploinsufficiency, because GATA3 is not expressed in heart or stomach.10 It is suggested that environmental or other genetic effects may be involved in the pathophysiology of a developmental abnormality in the heart.12
Since the GATA3 gene was first identified, 50 mutations have been described in patients with HDR syndrome [http://www.hgmd.org]. Upadhyay, et al.13 reported a de novo mutation rate of 45% in 40 patients with HDR syndrome. In this previous study, most GATA3 mutations were truncating mutations, including deletion (35%) and nonsense mutation (22.5%). The mutation identified in our patient involved a novel heterozygous mutation, c.153del (p.Phe51Leufs*144) in exon 1, which results in a frameshift with a premature stop codon, consequent truncating GATA3 protein or diminishing GATA3 mRNA due to mRNA decay. Furthermore, there has been no previous report on Korean HDR syndrome patient confirmed by molecular analysis.
In summary, this is the first case with HDR syndrome in Korea that was confirmed by genetic analysis, to involve a novel de novo GATA3 gene mutation. Further study is required to understand the functional and structural changes of proteins involved in this disorder and their associations with phenotypic spectrum.

 XML Download
XML Download