

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Radiation is an effective treatment modality that is commonly used to treat a variety of solid cancers. Despite many technical advances such as intensity modulation or tomotherapy, oral mucositis (OM) remains one of the most common and troubling side effects of radiation therapy. OM is especially prevalent in patients undergoing treatment for head and neck cancers, in which the oral cavity and oropharynx are included in the irradiation field.1 The vast majority of patients who receive cumulative radiation doses of more than 30 Gy to the oral mucosal field will develop this condition.2 The frequency of severe OM exceeds 90% in patients treated for cancers of the oral cavity and oropharynx.3 OM causes pain, dry mouth, and decreased appetite in patients, resulting in a decreased quality of life. OM affects treatment delivery and is often a dose-limiting factor that increases the morbidity, mortality, and cost of treatment.3 Despite various attempts to prevent and treat radiation-induced mucositis, no intervention modality has yet been identified that completely relieves OM.
The pathobiology of radiation-induced OM includes the release of free radicals, modified proteins, and proinflammatory cytokines such as interleukin-1β, prostaglandins, and tumor necrosis factor (TNF) by epithelial, endothelial, and connective tissue cells.4 These mediators cause further injury either directly or indirectly to the oral mucosa. Therefore, we sought a new antioxidant that could be helpful in preventing mucosal damage during the initial stages of radiation-induced OM.
We identified a candidate compound for preventing radiation-induced OM: the novel synthetic compound 3-amino-3-(4-fluoro-phenyl)-1H-quinoline-2,4-dione (KR22332). KR22332 was developed initially as a neuroprotective agent for ischemic stroke, epilepsy, and damage from chemical weapons such as organophosporus. It was found to exhibit antioxidant properties, and we showed previously that KR22332 exerts an otoprotective effect by preventing cisplatin-induced ototoxicity through the inhibition of reactive oxygen species (ROS) generation.5 Based on these findings, we felt that KR22332 could be a promising candidate protective agent against radiation-induced toxicity. In the present study, we evaluated the effectiveness of KR22332 as a protective agent against radiation-induced cellular toxicity in vitro.
MATERIALS AND METHODS
Synthesis and preparation of KR22332
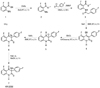
KR22332 was synthesized as described previously in five reaction steps using the commercially available starting material 2-amino-6-benzoic acid methyl ester (Fig. 1).5 The structure of KR22332 was identified by 1H nuclear magnetic reso-nance (NMR) spectroscopy: 1H NMR (300 MHz, CDCl3) δ 2.50 (br s, 2H, NH2), 6.76-6.80 (m, 2H, ArH), 6.95-7.01 (m, 2H, ArH), 7.36-7.44 (m, 3H, ArH), 9.87 (s, 1H, NH). The powdered KR22332 was prepared as solution with various concentrations using dimethyl sulfoxide (DMSO) (Santa Cruz Biotechnology, Dallas, TX, USA) as a solvent.
Cell culture
Human keratinocyte (HaCaT) cells were obtained from the American Type Culture Collection (Manassas, VA, USA), and the human head and neck squamous cell carcinoma (HNSCC) cell line HN3 was derived from patients with HNSCC. The cells were grown in high glucose Dulbecco's modified Eagle's medium (DMEM; Gibco, Grand Island, NY, USA) containing 10% fetal bovine serum (Gibco) and penicillin (50 U/mL) in a humidified atmosphere containing 5% CO2.
Irradiation
A single dose of radiation was delivered by opposed photon beams at a distance of 100 cm from the source to the axis using a 6 MV linear accelerator system (21EX Linear Accelerator; Varian Co., Palo Alto, CA, USA) as described previously.6
Cell viability assay
To determine cell viability, HaCaT and HN3 cells were seeded in 96-well plates at a density of 2×103 cells/well in 1 mL of complete medium after treatment with various concentrations of KR22322 (0-50 µg/mL) and various doses of radiation (0-20 Gy) or radiation plus KR22322. At 72 h after treatment, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide (MTT; Sigma Chemical Co., St. Louis, MO, USA) was added to 40 µL of the cell suspension for 4 h. After three washes with phosphate-buffered saline (PBS; pH 7.4), the insoluble formazan product was dissolved in dimethyl sulfoxide. The optical density of each culture well was measured using a microplate reader (Bio-Tek, Winooski, VT, USA) at 560 nm.
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP-biotin nick end labeling (TUNEL) assay
HaCaT cell apoptosis was determined by the TUNEL method using an in situ cell detection kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer's instructions. The cells, prepared as described previously,5 were exposed to medium with KR22332 (10 µg/mL) only, radiation only (8 Gy), or radiation (8 Gy) plus KR22332 (10 µg/mL) for 24 h. The stained cells were analyzed under a fluorescence microscope (400×; Carl Zeiss, Oberkochen, Germany); the number of TUNEL-positive cells was determined in triplicate.
Annexin V-fluorescein isothiocynate (FITC)/propidium iodide (PI) double staining
A quantitative analysis of apoptotic cell death caused by radiation was performed using a FITC Annexin V Apoptosis Detection kit II (Becton Dickinson, Franklin Lakes, NJ, USA). Cells were stained using an Annexin V-FITC apoptosis detection kit, following the manufacturer's protocol. Briefly, HaCaT cells were plated at 3×105 cells/well in a 6-well plate, incubated for 24 h, and then treated with KR22332 (10 µg/mL) only, radiation only (8 Gy), or radiation (8 Gy) plus KR22332 (10 µg/mL) for 72 h. The cells were then harvested, washed with cold PBS, and subjected to Annexin V-FITC and PI staining in binding buffer at room temperature for 10 min in the dark. The stained cells were analyzed by fluorescence-activated cell sorting (FACS ARIA3; BD Biosciences, San Jose, CA, USA) using WinMDI 2.9 software.
Mitochondrial membrane potential (MMP) assay
The MMP of intact cells was measured by flow cytometry with the lipophilic cationic probe 5,5 V,6,6 V-tetrachloro-1,1 V 3,3 V-tetra ethylbenzimidazol-carbocyanine iodide (JC-1; Molecular Probes, Eugene, OR, USA). Briefly, the culture medium was removed from adherent HaCaT cells, and the cells were rinsed with PBS. The cell monolayers were then incubated with DMEM and 5 mg/mL JC-1 at 33℃ for 20 min. The cells were subsequently washed twice with cold PBS, trypsinized, and then treated with KR22332 (5 or 10 µg/mL) only, radiation only (8 Gy), or radiation (8 Gy) plus KR22332 (5 or 10 µg/mL) for 24 h. The pelleted cells were then resuspended in 500 µL of PBS. The change in MMP was measured by flow cytometry.
Measurement of intracellular ROS generation
Intracellular generation of ROS was quantified using 5-(and 6)-carboxyl-20,70-dichlorodihydro fluorescein diacetate (DCFDA; Molecular Probes). For the assay, HaCaT cells were cultured overnight on 6-well plates and then exposed to radiation (8 Gy) in the presence or absence of KR22332 (10 µg/mL). After radiation, the cells were incubated for 24 h and then treated with 10 µM DCFDA in serum-free medium for 10 min at 33℃ in the dark. The oxidative burst (hydrogen peroxide) was detected using a FACScan flow cytometer (Becton Dickinson) with excitation and emission settings at 488 and 530 nm, respectively.
Western blot assay
Total proteins were extracted using radio immunoprecipitation assay (RIPA) buffer (Sigma-Aldrich, St. Louis, MO, USA), following the manufacturer's instructions. Protein concentrations were measured using the Bio-Rad Dc Protein Assay (Bio-Rad Laboratories, Hercules, CA, USA). The proteins were separated by electrophoresis as described previously. The following antibodies were used for Western blot analysis: anti-ataxia telangiectasia mutated protein (ATM), anti-p53 (Ser 15, 20, and 46), anti-TNF-α, anti-cleaved caspase-3, and anti-PARP (1:1000; Cell Signaling Technology, Danvers, MA, USA).
Statistical analyses
Student's t-test was used for statistical analyses of the data. All statistical analyses were conducted using SPSS 20.0 statistical software (SPSS Inc., Chicago, IL, USA). Parameters of the data from three independent experiments are expressed as mean±SD. p<0.05 was considered to be significant (*p<0.05; **p<0.01; ***p<0.001).
RESULTS
KR22332 increases HaCaT cell viability after radiation treatment
As shown in Fig. 2A, radiation decreased HaCaT cell viability in an intensity-dependent manner. KR22332 did not show significant toxic effects on the cells until 10 µg/mL (Fig. 2B). Irradiated HaCaT cells treated with various concentrations of KR22332 (0, 1, 10, and 50 µg/mL) were significantly protected from radiation-induced cytotoxicity in a dose-dependent manner (Fig. 2C). The slight decrease in cell viability observed in the 50 µg/mL KR22332-treated group might be due to a cytotoxic effect of the high dose of KR22332 (Fig. 2B). HaCaT cells displayed optimal viability at 10 µg/mL of KR22332 after irradiation (8 Gy). Therefore, the remaining experiments were performed using 10 µg/mL of KR22332 and 8 Gy of irradiation.
KR22332 does not decrease the therapeutic effect of radiation against cancer cells
If KR22332 decreases the effect of anti-cancer treatment, KR22332 might not be applied in clinical setting in spite of protective property on radiation induced mucosal damage. Therefore, we investigated the effect of KR22332 on the therapeutic effect of radiation against cancer cells derived from HNSCC. Interestingly, differing from the normal keratinocyte HaCaT cells (Fig. 2B), KR22332 alone showed decrease of cancer cell viability (Fig. 3A). As shown in Fig. 3B, irradiation significantly decreased the viability of HN3 cells (p<0.001). However, KR22332 did not show any significant protective effect on the viability of HN3 cells. The results indicated that KR22332 did not decrease the effect of radiation treatment on the cancer cells while it showed protective effect on the normal epithelial cell from radiation induced damage.
KR22332 protects against radiation-induced HaCaT cell apoptosis
A TUNEL assay and 4',6-diamidino-2-phenylindole (DAPI) staining were performed to determine whether radiation-induced cell death in HaCaT cells occurred by apoptosis, and whether this cell death could be prevented by KR22332 treatment. As shown in Fig. 4A, irradiation increased the number of TUNEL-positive cells while KR22332 treatment obviously decreased the number of TUNEL-positive cells. Moreover, DAPI staining revealed that KR22332 treatment decreased the radiation-induced nuclear condensation, DNA fragmentation, and perinuclear apoptotic bodies caused by irradiation in HaCaT cells.
We used flow cytometry to quantify and verify the number of apoptotic HaCaT cells induced by radiation treatment. Annexin V-FITC/PI staining was used to analyze the percentage of apoptotic cells after irradiation in the absence or presence of KR22332 (Fig. 4B). The number of late apoptotic cells is shown in the upper right quadrant; and the number of early apoptotic cells is plotted in the lower right quadrant of the histograms. We found that radiation plus KR22332 treatment (10 µg/mL) significantly decreased the number of total apoptotic cells (mean, 20.0%) compared to cells treated with radiation alone (mean, 37.4%; p<0.05). HaCaT cells treated with KR22332 alone showed no significant change in the number of Annexin V-FITC/PI-positive cells compared to the control. Taken together, these results demonstrate that irradiation promoted apoptotic cell death in HaCaT cells, but this effect was inhibited by KR22332.
KR22332 protects against radiation-induced decrease of MMP
The MMP can be used as an index of mitochondrial pore opening, which is an indicator of mitochondrial dysfunction. The quantitative analysis of red and green fluorescent signals from JC-1 reflects the degree of mitochondrial damage. We examined the effect of radiation on the MMP in HaCaT cells to determine whether the loss of MMP could play a role in radiation-induced apoptosis. As shown in Fig. 5A, a high MMP was maintained in the control cells, as indicated by predominantly red fluorescence of the JC-1 dye. However, radiation treatment increased the degree of green cell fluorescence, indicating a loss of MMP and mitochondrial damage. KR22332 treatment (5 and 10 µg/mL) significantly ameliorated reduced the green fluorescence caused by irradiation, and the red fluorescence was significantly restored (p<0.05 and p<0.05, respectively) (Fig. 5), indicating that KR22332 attenuated radiation-induced changes in the MMP.
KR22332 inhibits intracellular ROS generation induced by radiation
We next investigated the effect of radiation on the intracellular generation of ROS. Cells were treated with 8 Gy of radiation, and the level of intracellular ROS was monitored by flow cytometry with the peroxide-sensitive fluorescent probe DCFDA. Irradiation significantly increased the generation of intracellular ROS (p<0.05). In contrast, KR22332 significantly inhibited radiation-induced ROS generation in a dose-dependent manner. KR22332 inhibited radiation-induced ROS generation as much as N-acetyl-L-cysteine (NAC; 10 mM), a potent ROS scavenger (Fig. 6). These results suggest that KR22332 protects HaCaT cells against radiation-induced toxicity by reducing intracellular ROS formation.
KR22332 rescues HaCaT cells by inhibiting the caspase-3 pathway and suppressing the expression of ATM, p53, and TNF-α
To elucidate the underlying mechanisms of radiation-induced cell death and the protective effect of KR22332 in HaCaT cells, we evaluated the genes related to apoptosis such as cleaved caspase-3, PARP, ATM, p53 (Ser 15, 20, and 46), and TNF-α. As shown in Fig. 7, radiation increased the expression of cleaved caspase-3 and PARP cleavage, and KR22332 cotreatment inhibited their expression. Moreover, KR22332 inhibited the expressions of ATM, P-p53 (Ser 15 and 46), and TNF-α, which were significantly increased in cells treated with radiation only. These results suggest that KR22332 blocked radiation-induced apoptosis via down-regulation of ATM-p53 and TNF-α signaling.
DISCUSSION
OM remains one of the most common and troubling side effects of standard chemoradiation regimens used for the treatment of head and neck cancers. Patients undergoing radiotherapy often suffer from altered taste, pain, dry mouth, decreased appetite, and ulcers owing to OM, which results in a significantly decreased quality of life and prognosis.7 Despite many advances aimed at treating or preventing OM, the treatment options remain limited.8 Therefore, novel agents to prevent and mitigate the development of OM in patients during head and neck cancer treatment are clearly needed.
In this study, we used the human keratinocyte HaCaT cell line as an in vitro model of the human oral mucosa to investigate the possible OM-protective effects of newly developed compound KR22332. Although additional cell populations such as fibroblasts and endothelial cells make up mucosal tissue, keratinocytes are among the first cells to encounter environmental hazards;9 human keratinocyte cultures are often used to investigate the effects of ionizing radiation.10 Tobita, et al.11 recently developed an in vitro model of OM using HaCaT human keratinocytes, and Donetti, et al.9 used human keratinocytes as an in vitro model of oral mucosa to study the early effects of radiation-induced OM. Likewise, we used HaCaT cells as an in vitro model of the human oral mucosa; however, a major shortcoming of our model is the lack of a vascular response and the influence of the immune system.
Historically, the pathobiology of OM has been described as the indiscriminate destruction of rapidly dividing basal epithelial stem cells by radiation or chemotherapy. However, the current concept is that OM results from a complex sequence of biological events, including the release of free radicals (e.g., ROS), modified proteins, and pro-inflammatory cytokines (e.g., interleukin-1β, prostaglandins, and TNF-α) by epithelial, endothelial, and connective tissue cells, rather than being the direct result of clonogenic cell death.12 KR22332 was developed recently as a novel neuroprotective drug to treat ischemic stroke, epilepsy, or exposure to chemical weapons such as organophospates. In fact, we found that KR22332 has some antioxidant properties.5 We demonstrated previously that radiation-induced cell death in human keratinocyte HaCaT cells occurred by apoptosis related to increased membrane damage and the generation of intracellular ROS.6 Therefore, we postulated that the novel antioxidant KR22332 could be a candidate protective agent for radiation-induced OM. As hypothesized, our TUNEL assay and Annexin-V/PI flow cytometric results revealed that KR22332 prevented radiation-induced apoptosis in HaCaT cells. Moreover, KR22332 decreased ROS generation and prevented instability in the MMP, which were induced by irradiation, suggesting that the radioprotective capacity of KR22332 is derived from the inhibition of ROS generation and preservation of mitochondrial integrity. A Western blot analysis revealed that KR22332 reduced the phosphorylation of ATM, p53, and TNF-α in response to irradiation. These results are in support of our previous study, in which we suggested that the phosphorylation level of ATM and p53 signaling is related to radiation-induced apoptosis.6
ATM is activated by DNA double-strand breaks and regulates apoptosis via p53,13 and the inhibition of ATM blocks the DNA damage response and apoptosis in normal resting cells.14 We recently showed that ATM/p53-dependent NSAID-activated gene-1 (NAG1) expression induced apoptosis in cells.15 Furthermore, the activation of ATM and p53 is known to be mediated by ROS.16 In the present study, we examined the activation state of the ATM-p53 apoptotic pathway in HaCaT cells with or without KR22332 treatment. Indeed, KR22332 reduced apoptosis, and the expressions of ATM and p53, and ROS production (all of which were increased by irradiation) in HaCaT cells.
TNF-α is a potent cytokine that functions in inflammation, cell proliferation, and apoptosis; and TNF-α-induced apoptosis has been well documented.17 There are two main pathways in TNF-α signaling: a pro-survival/pro-inflammatory pathway that activates; nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kB), mitogen-activated protein kinase (MAPK), and a proapoptotic pathway in which ROS, a caspase cascade, and mitochondria function as downstream mediators.18 Curra, et al.19 reported that the inhibition of TNF-α is associated with OM in vivo. Our results suggest that TNF-α is related to radiation-induced apoptosis in HaCaT cells, and that the protective capacity of KR22332 against radiation-induced ROS generation and apoptosis is related to the inhibition of TNF-α.
ROS are generated under many stressful conditions, and they play an important role in cell survival and cell death by apoptosis or necrosis.20 Intracellular ROS serve as major mediators of radiation-induced cellular damage. When ROS generation exceeds cellular antioxidant defenses, cell damage ensues.16,20 It is well established that ROS have an important function in cell survival and cell death related with ATM-p5316 or TNF-α signaling.18 The present results also suggest that irradiation induced apoptosis in human keratinocytes in vitro, and that KR22332 exhibited a cytoprotective property as a scavenger of ROS by inhibiting the ATM-p53 and TNF-α pathways. To our knowledge, this is the first report to suggest that both the ATM-p53 and TNF-α signaling pathways are involved in the pathogenesis of radiation-induced mucositis. In conclusion, we demonstrated that the novel synthetic compound KR22332 protects HaCaT cells from radiation-induced damage and death by preventing changes in the MMP and generation of ROS, and by inhibiting activation of the ATM and p53 pathways and TNF-α-mediated apoptotic signals. This effect was confirmed using an in vivo zebrafish model. HaCaT cells are an accepted in vitro model of oral epithelial cells; therefore, these findings suggest the potential of KR22332 in the reduction of radiation-induced OM, which is a common complication of chemoradiation treatment for head and neck cancers.






 XML Download
XML Download