

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Adrenoleukodystrophy (ALD, OMIM# 300100) is a progressive neurodegenerative disease characterized by an accumulation of very long chain fatty acids (VLCFA).1,2 Alterations in the ATP-binding cassette subfamily D, member 1 (ABCD1) gene result in dysfunction of the ALD protein, which participates in the peroxisomal degradation of VLCFAs. The accumulation of VLCFAs primarily affects the central nervous system, adrenal cortex and testis; nevertheless, its clinical presentation varies greatly.3 Cerebral ALD and adrenomyeloneuropathy (AMN) are the main phenotypes of ALD, but these two phenotypes revealed distinct clinical and pathologic features. Cerebral ALD is usually diagnosed at a young age and is the most severe phenotype, characterized by rapidly progressive inflammatory cerebral demyelination. AMN usually appears between the ages of 20 and 50 years and presents as a slowly progressive non-inflammatory axonopathy that mainly involves the spinal cord and peripheral nerves. However, about 20% of AMN patients exhibit cerebral demyelinating involvement later in life.4 This risk is associated with early onset, usually between 20 and 35 years of age, and decreases after the age of 45 years.5 In addition to the main phenotypes, Addison-only, spinocerebellar (olivo-ponto-cerebellar) and asymptomatic phenotypes are occasionally reported.6 Thus, this heterogeneity in clinical presentations makes it difficult to diagnose ALD. The clinical characteristics of adult-onset ALD in Koreans have not yet been evaluated, and only a few cases have been reported.7,8,9,10,11 Therefore, the present study was designed to investigate the characteristics of clinical, electrophysiological, radiological and genetic data in Korean AMN patients.
MATERIALS AND METHODS
Patients
From January 2006 to June 2013, we retrospectively enrolled Korean AMN patients treated at four hospitals: Severance Hospital, Yonsei University College of Medicine; Mokdong Hospital, Ewha Womans University School of Medicine; Kangbuk Samsung Hospital, Sungkyunkwan University School of Medicine; and the hospital of National Cancer Center. For patient selection, patients who fulfilled the two following conditions were selected among medical records: 1) patients with spastic paraparesis and 2) patients with increased plasma VLCFA levels, especially the C26:0/C22:0 and C24:0/C22:0 ratios. Based on these criteria, we identified twelve AMN patients. Nine patients were evaluated at Severance Hospital, one was evaluated at Mokdong Hospital, one was evaluated at Kangbuk Samsung Hospital, and one was evaluated at the hospital of National Cancer Center. Patient 7 was previously reported on in a Korean journal.9 The present study was initiated after obtaining approval from the Institutional Review Board at our institution (IRB Number: 4-2013-0167). Written informed consent was exempted by the board because this was a retrospective study.
Methods
Clinical data were collected from medical records and included age of symptom onset, disease duration, family history, cognition, cranial nerve function, motor function, sensory disturbance, cerebellar function, deep tendon reflex and other associated signs, such as urinary disturbances, hypoglycemia, low blood pressure and increased skin pigmentation. Plasma adrenocorticotropic hormone (ACTH) and cortisol concentrations were measured in seven patients. Adrenal function was defined as abnormal if the baseline cortisol level was less than 10 µg/dL and the baseline plasma ACTH level was greater than 100 pg/mL, or if the cortisol response to ACTH stimulation test was suboptimal. Additionally, data were collected to assess damage to the nervous system using electrophysiological and radiological studies. Electrophysiological studies were performed using standardized techniques at room temperature. Motor and sensory nerve conduction studies were tested in eleven patients. Visual evoked potential tests were performed in seven patients. Moreover, median and posterior tibial nerve somatosensory evoked potential tests were performed in eight and 12 patients, respectively. Radiological data included spinal and brain MRI scans, which were performed in 12 and 10 patients, respectively. Mutations were identified by direct sequencing using a primer set that covered all 10 exons and flanking intron regions of the ABCD1 gene as described by Boehm, et al.12 The ABCD1 gene mutations were analyzed in eleven patients.
RESULTS
Clinical analysis
The clinical spectrum of the patients is summarized in Table 1. All 12 patients were men and exhibited increased plasma VLCFA levels (Supplementary Table 1, only online). Patient ages ranged between 19 and 57 years, and age at symptom onset ranged between 18 and 55 years. Disease duration ranged between 6 months and 7 years. Family history was positive in two patients (patient 10 and 12): the nephew on the mother's side of patient 10 was diagnosed with Addison-only phenotype at the age of seven years. Additionally, even though the mother of patient 12 had not complained of any muscle weakness and sensory disturbance, clinical examination revealed subtle spastic paraparesis and decreased proprioception on her lower extremities. However, we could not confirm her disease due to her refusal.
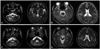
Onset symptoms included lower limb weakness in eleven patients and dysarthria in one patient (patient 10). The phenotype distributions consisted of AMN without cerebral involvement in seven patients, AMN with cerebral involvement in two patients, and the spinocerebellar phenotype in three patients. Among them, three patients (patients 8, 10, and 12) were initially misdiagnosed with multiple sclerosis, unknown brainstem encephalopathy and spinocerebellar ataxia, respectively. This was because patient 8 exhibited unilateral lesions in the right cerebellar and temporo-occipital regions on brain MRI scans (Fig. 1A), patient 10 mainly complained of dysarthria, and patient 12 presented with prominent cerebellar limb ataxia.
At clinical examination, all 12 patients showed some degree of muscle weakness, increased tendon reflexes, and sensory disturbance on the lower extremities. Eight patients had urinary disturbance (urgency, retention or incontinence). Among two patients with cerebral involvement, patient 8 demonstrated personality change, visual disturbance and dysarthria. Patient 9 reported visual disturbance. Three patients with cerebellar ataxia exhibited limb ataxia predominantly in the unilateral arm, even though the affected arms did not show any sensory impairment. In addition to cerebellar ataxia, patient 10 had memory impairment and dysarthria, and patient 12 had dysarthria.
In addition to neurologic deficits, two patients (patients 8 and 11) reported thin and scanty scalp hair since adolescence. Symptoms of adrenal insufficiency such as increased skin pigmentation, hypoglycemia and low blood pressure were not described in any of the patients. However, measurement of plasma cortisol and ACTH concentration, as well as ACTH stimulation test, revealed abnormal responses in five of seven tested patients.
Table 2 lists the electrophysiological and radiological features of twelve Korean AMN patients. Nerve conduction studies demonstrated axonal sensorimotor polyneuropathy in four of 11 tested patients. Visual evoked potential tests revealed abnormalities in three of seven tested patients. Median and posterior tibial somatosensory evoked potential tests demonstrated central conduction defects in all tested patients. On radiological studies, spinal MRI scans revealed diffuse cord atrophy and subtle T2 hyperintensity in 10 and two, respectively. However, Brain MRI scans revealed parenchymal abnormalities in six of 10 tested patients. These brain abnormalities reflected the clinical phenotypes. Among seven AMN patients without cerebral involvement, three patients showed only T2 high signal intensities in the corticospinal tract. Among two patients with cerebral involvement, patient 8 showed a lesion involving the corticospinal tract, right temporo-occipital subcortex and cerebellum with patchy enhancement (Fig. 1A). Patient 9 presented with T2 high signal intensities in not only the corticospinal tract but also the splenium (Fig. 1B). Among three patients with the spinocerebellar phenotype, patient 10 was presented with T2 hyperintense lesions around the dentate nuclei (Fig. 1C). However, even though patient 12 had cerebellar ataxia, brain MRI scan revealed only a lesion in the corticospinal tract (Fig. 1D).
Mutational analysis
We identified nine different mutations of the ABCD1 gene in 10 of 11 tested patients, but no mutation was found in one (Table 3, Fig. 2). Among the nine different mutations, five (56%) were missense, two (22%) were frameshift, and two (22%) were deletion. Additionally, mutations were identified in two hot spots: the transmembrane domain (four mutations, 44%) and the nucleotide binding domain (three mutations, 30%). Among nine different mutations, eight were previously reported in the ALD database (http://www.x-ald.nl/).6,13,14,15 However, they were associated with not only AMN but also childhood cerebral ALD, adult-onset cerebral ALD, and the asymptomatic phenotype. Additionally, even though patients 6 and 8 had the same genotype (c.1679C>T), one did not show cerebral involvement and the other showed clinical and radiological involvement in the brain. We identified one novel duplication (c.277_296dup20) mutation in the present study.
DISCUSSION
The present study reported the clinical, mutational, electrophysiological and radiological aspects of 12 Korean AMN patients. Our results were compatible with previously documented findings of AMN.
It is well known that the principle biochemical abnormality of ALD is an excess of VLCFAs. However, the pathogenesis and pathophysiology of ALD is very complex and not yet completely understood. Nevertheless, neuropathologic study has revealed a fundamental difference between AMN and cerebral ALD.16 Additionally, studies have shown that the clinical phenotypes thereof are not correlated with genotype, even in members of the same family with the same mutation.17,18 This suggests that beyond mutations in the ABCD1 gene, other genetic, epigenetic or environmental factors might be associated with the clinical presentation of ALD. For the evaluation of nervous system involvement in AMN patients, it is known that electrophysiological studies, especially somatosensory evoked potential testing, are powerful modalities.19,20,21 In regards to radiological study, spinal MRI scans usually show nonspecific cord atrophy in AMN patients.22 However, brain MRI scan occasionally shows abnormal parenchymal lesions. Commonly, a lesion in the corticospinal tract, which probably reflects Wallerian degeneration, is found in AMN patients.22 Other demyelinating lesions are considered indicative of progression.
The present study reports that Korean AMN patients show various clinical presentations from spastic paraparesis to cortical and cerebellar involvement, and is compatible with previous studies, except for a large proportion of spinocerebellar phenotype patients. The spinocerebellar phenotype is characterized by clinical presentations of spinocerebellar ataxia, and is also referred to as the olivo-ponto-cerebellar phenotype. The pathophysiology of the spinocerebellar phenotype is not known. However, these patients frequently show white matter lesions of the cerebellum and brainstem on brain MRI scans, suggesting that the clinical presentation of the spinocerebellar phenotype is probably associated with involvement of the dentate-rubro-thalamo-cortical tract.23,24,25,26,27 Actually, patient 10 who presented with spinocerebellar phenotype also demonstrated a lesion of the bilateral dentate nuclei in the present study. The spinocerebellar phenotype is very rare and affects only 1-2% of all ALD patients in western countries.6 However, this rare phenotype is observed relatively frequently in Japan.1,6,28 Therefore, common modulating factors between Japan and Korean ALD patients may be present.
Electrophysiological and radiological features in our study were compatible with previously documented findings, as well. In the present study, brain MRI findings provided further information on clinical phenotype and prognosis. Among three patients with spinocerebellar phenotype, patient 10 comprised a lesion in dentate nucleus. Moreover, two patients with clinical cerebral involvements showed lesions in the splenium and a temporo-occipital subcortical lesion. Previous studies reported that parieto-occipital subcortical lesions with contrast enhancement were especially associated with rapid progression.22,29 Based on previous studies, patient 8 may be at high risk of rapid progression.
The mutational analysis in our study identified one novel mutation. The eight other mutations had been previously reported and were associated with not only AMN phenotypes but also various other phenotypes. In the present study, patients 6 and 8 had the same genotype, but exhibited different phenotypes. Additionally, patient 10 reported interfamilial clinical differences. This was compatible with previous results for a lack of correlation between genotype and phenotype.17,18 Furthermore, the presence of mutational hotspots and the predominance of missense variants were similar to previous reports.5,6
A number of important limitations in our study need to be considered. First, the main limitation of our study was the relatively small number of patients. Therefore, further studies are needed to verify the consistency of our result in larger cohorts. Second, our study was retrospectively designed based on chart reviews. Third, we usually performed VLCFA analysis on men with spastic paraparesis. We probably missed many ALD patients such as symptomatic female carriers of ALD and patients with only endocrinological abnormalities. Moreover, the mother of patient 12 was thought to be a symptomatic female carrier but she refused further evaluation. The nephew of patient 10 was diagnosed with an Addison-only phenotype. However, we were unable to obtain his medical records. Nevertheless, we did not include these two patients in the present study. Fourth, the pathogenic mutation was not identified in patient 5. As patient 5 showed elevated plasma VLCFA levels with typical clinical features of ALD, he was diagnosed with AMN biochemically.29 There are two possibilities for the inability to identify the mutation. One explanation could be that patient 5 had large deletions of one or more exons that could not be identified by direct sequencing. Another possibility is that the mutational analysis failed to identify alterations of the ABCD1 gene due to the presence of ABCD1 paralogs on chromosome 2q11, 10p11, 16p11 and 22q11.12. Actually, we were unable to identify the mutation of patient 12 in the first genetic test, as well. However, a c.901-1G>A mutation was identified after repetitive tests and change of primers.
In conclusion, the present study is the first to report on the clinical and mutational spectrum of Korean AMN patients, and confirms various clinical presentations and the usefulness of brain MRI scan.



 XML Download
XML Download