

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Short stature is one of the most common causes of referral to pediatric endocrinologist. It is defined as a condition when height is more than 2 standard deviation score (SDS) below the corresponding mean height for a given age, gender, and a population group. Although the causes of short stature have been identified in some children, a large number of such children remain without a definitive diagnosis and are considered as having idiopathic short stature (ISS). Therefore, ISS can be diagnosed after excluding the evidence of systemic, endocrine, nutritional, or chromosomal abnormalities at the end of the diagnostic evaluation.1 ISS represents a heterogeneous group of children having many unidentified causes of short stature, such as constitutional delay in growth and puberty, familial short stature, neurosecretory dysfunction, and disorders with gene mutation for molecules involved in growth process in any level including bio-inactive growth hormone (GH), partial GH insensitivity, GH receptor signaling defects, mutations of the insulin-like growth factor (IGF)-I or IGF-I receptor, and short stature homeobox-containing gene (SHOX) haploinsufficiency. It is estimated that approximately 60-80% of all short children have a diagnosis consistent with ISS.2
Recombinant human GH has been used since the approval by US Food and Drug Administration (FDA) to use in children with growth failure due to GH deficiency in 1985. The indications for GH treatment have gradually extended from GH deficiency to non-GH deficient short stature, such as chronic renal insufficiency, Turner syndrome, Prader-Willi syndrome, short children born small for gestational age (SGA), ISS, and Noonan syndrome.3 GH treatment for children with ISS was approved in the United States in 2003. Such an approval of GH treatment for ISS was based on the efficacy results obtained from the randomized studies, suggesting that GH treatment tends to increase the adult height of children with ISS by about 4-6 cm.4-6 They recommended GH treatment (at doses up to 53 µg/kg·d) for children with ISS shorter than -2.25 SDS (1.2 percentiles), associated with height velocity unlikely to permit attainment of adult height in the normal range. More recently, a consensus statement of the Growth Hormone Research Society, the Lawson Wilkins Pediatric Endocrine Society, and the European Society for Pediatric Endocrinology proposed that children with ISS whose heights were less than -2 to -3 SDS warranted consideration for treatment.7 The consensus statement also reported that GH therapy can increase the adult height in children with ISS by 3.5-7.5 cm (mean treatment of 4-7 years), and has a similar safety profile to other GH indications.7
The aim of this study was to investigate the efficacy and safety of short-term GH treatment in prepubertal children with ISS to evaluate whether short-term GH therapy was efficacious for increasing height SDS without any adverse effects.
MATERIALS AND METHODS
Subjects
Short children with ISS whose chronological age (CA) was 5 years or older, with a corresponding bone age (BA) of no more than 10 years in girls and no more than 12 years in boys were included. Inclusion criteria were short stature, defined as height less than 3rd percentile for age and gender according to the Korean population-based reference;8 peak GH responses using one of three GH stimulation tests (clonidine, insulin, or L-dopa) greater than 7 ng/mL (considered a normal GH level in children); prepubertal stage according to Tanner stage criteria based on breast development in girls and testicular volume in boys; no previous GH treatment at least within 6 months; children with normal thyroid hormone level (normal thyroid hormone level through hormone therapy inclusive); no significant chronic diseases, chromosomal abnormalities, skeletal dysplasia, dysmorphic syndromes, short children born small for gestational age, and taking any drug suspected to interfere with GH secretion or action. Children on hormone therapy such as estrogen, androgen, anabolic steroid, corticosteroid, and methylphenidate that could interfere with growth were excluded from the study. In total, 36 children with ISS were enrolled into the study, and 3 children were not included for the efficacy analysis, primarily owing to protocol violations. Data from 33 children with at least one available treatment efficacy data were included in the efficacy analysis (full analysis set, FAS). The FAS was defined as dataset from those who received at least one dose of the study drug and who had at least one post-dose auxological measurement. Children with major protocol violations, such as poor study drug compliance (less than 70%), violations of eligibility criteria, and pubertal advance during the study were excluded from FAS. Per-protocol set (PPS) comprised of 25 children who had followed most of the protocol procedures was the main population for efficacy presentation. Safety data analyses were performed on a safety set that comprised of data from all randomized patients who received at least one dose of the study drug. The study protocol was approved by the Ethical Committees of the participating institutions and Korean FDA. Informed consents were obtained from parents or legal guardians of each child and from all appropriately aged children.
Methods
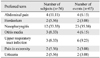
This study was an open-label, multicenter, interventional study. After evaluating growth status during a 6-month pre-study period, eligible children were given subcutaneous injections of GH with a dose of 0.37 mg/kg/week for 26 weeks, dividing equally to be given for 6 consecutive days per week. GH (Eutropin®) was provided by LG Life Sciences Ltd. (Seoul, Korea). Subjects were required to visit the investigational site three times including baseline, week 13 and week 26 to measure height, weight, IGF-I, IGF binding protein (IGFBP)-3, thyroid function, hemoglobin A1C and other laboratory test items. Peak plasma GH levels after GH stimulation test using insulin, clonidine, or L-dopa ranged from 14.82 ng/mL to 22.52 ng/mL (Table 1). Pubertal stage was also evaluated by pediatric endocrinologist at every visit. The measurements of X-ray of left wrist for BA determination and GH antibody were performed both at baseline and week 26. BA was determined in one investigational site by Greulich-Pyle method. Telephone calls were made by investigational site staff or investigators at week 5 and week 18 to ensure treatment adherence and to investigate the adverse events.
Two outcome variables were used for efficacy analyses: 1) mean change in height velocity at week 13 and week 26 during 6-month treatment with GH; 2) gain in height SDS at week 13 and week 26 during 6-month treatment with GH. Annualized height velocities are expressed as centimeters per year, computed as the change in height (centimeters) divided by the change in age (years). The height SDS was calculated using the growth standard for Korean children and adolescents.8
Adverse event rates were summarized by System Organ Class MedDRA (a registered trademark of the International Federation of Pharmaceutical Manufacturers Associations) thesaurus terminology (http://www.meddramsso.com). Investigators were instructed to report any adverse clinical or laboratory findings when there was a suspicion that GH treatment may have been related to the adverse events.
Statistical analyses
The statistical analyses were performed using the standard statistical package SPSS, version 15.0 (SPSS Inc., Chicago, IL, USA). Results were expressed as mean±SD unless otherwise specified. Descriptive statistics were used to summarize baseline characteristics, adverse events, and laboratory results. Efficacy analyses were performed using paired t-test or Wilcoxon's signed rank test to detect the difference from baseline. Differences in laboratory results from baseline were determined using paired t-test or Wilcoxon's signed rank test. Differences in laboratory values, separately analyzed in normal and abnormal range group, were determined using McNemar's test. We considered a p value less than 0.05 to be statistically significant.
RESULTS
Baseline characteristics
Total of 36 subjects with confirmed ISS were enrolled in the study, and 33 subjects were included in the FAS and 25 in the PPS (Fig. 1). All of the enrolled subjects who received at least one dose of GH were included in the safety analysis. Baseline demographics of FAS and PPS are presented in Table 1. Both genders were evenly distributed and subjects' age ranged from 5 to 12 years. Mean BA was delayed about one year, compared to mean CA. Only one subject had previous history of GH treatment in the FAS while PPS comprised of only treatment naïve subjects. Height SDS for CA was -2.35±0.53 in the FAS and -2.39±0.54 in the PPS.
Efficacy results
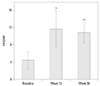
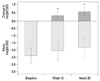
Mean growth velocities at baseline, week 13, and week 26 were 4.49±1.98, 11.67±4.20, and 10.85±2.34 cm/yr, respectively. After 26 weeks of treatment with GH, significant increase in height velocity by 6.36±3.36 cm/year, compared to the baseline value, was observed (p<0.001) (Fig. 2, Table 2). The gain in height SDS was 0.33±0.23 at week 13 (p<0.0001) and 0.57±0.27 at week 26 (p<0.0001) (Fig. 3). These results demonstrated the rapid growth enhancement during GH treatment, indicating that even the short-term GH treatment can have stimulatory effect on growth. BA also increased after treatment and the mean difference at week 26, compared to baseline, was 0.31±0.38 yr, which was a significant change (p=0.0005) (Table 2). However, BA was not too advanced for CA as bone maturation rate (change in BA/change in CA) was 0.58±0.72. Serum IGF-I level almost doubled at week 26 compared to baseline (150.4±67.8 ng/mL at baseline vs. 290.0±110.4 ng/mL at week 26; p<0.0001). Serum IGFBP-3 level also significantly increased after treatment of GH (2442.9±521.8 ng/mL at baseline vs. 3368.8±685.6 ng/mL at week 26; p<0.0001)(Table 2). Those subjects whose IGF-I or IGFBP-3 levels at baseline were less than the lower limit of normal range achieved normal levels of IGF-I and IGFBP-3 at week 26. Analyses using FAS on these efficacy parameters yielded similar results (data not shown).
Safety results
Among total 36 subjects, 23 (63.89%) children reported 65 adverse events (Table 3 and 4). The most common adverse event was nasopharyngitis (35.38%, 23 events among 65 events), which was reported in 12 children, and it was a frequently expected adverse event in the study population. All the remaining adverse events were mild in intensity. Mild pruritus on injection site that was reported twice in a same subject was assessed as being related to the study drug and it was resolved without any intervention. No serious adverse events were reported and no withdrawals due to adverse events. No clinically significant changes in laboratory values were observed except overall increase in alkaline phosphatase (308.08±202.54 IU/L at baseline vs. 436.26±290.60 IU/L at week 26; p<0.0001), which was considered to be associated with bone growth after administration of GH (Table 2). Statistically significant increase in the number of children with abnormally high density lipoprotein at week 13 was observed [8 (22%) children at baseline vs. 13 (41%) children at week 13; p=0.0253], however, this change was not clinically significant as other lipid profiles were within normal ranges (Table 2). All 35 children tested for GH antibody at baseline showed negative responses, while 3 (9.4%) children out of 32 tested for antibody at week 26 showed positive responses without deteriorating effect on growth (Table 2).
DISCUSSION
In the present study, we showed that the daily administration of GH in prepubertal children with ISS resulted in a marked increase in the mean height velocity during the 6 month-treatment period. Yearly height velocity increased by 6.36 cm/yr after GH treatment (from 4.49 cm/yr at baseline to 10.85 cm/yr at week 26). The accelerated height velocity resulted in significant increase in height SDS for CA by 0.57±0.27 during 6-month treatment (from -2.39 at baseline to -1.82 at week 26). Moreover, height SDS for BA was also increased during the treatment, suggesting that GH treatment might lead to improved ultimate stature without inducing advanced bone maturation. Proper bone maturation after GH treatment with a dose of 0.37 mg/kg/week was also supported by the finding that bone maturation rate was 0.58±0.72.
Recent reviews about the results of GH therapy for ISS showed that GH treatment can increase short-term growth and improve final adult height.9,10 Several clinical trials have reported that GH treatment for ISS increases height velocity in the first year, followed by gradual tapering off in subsequent years. Overall, mean height velocity increased from 4-5 cm/year before treatment to 8-9 cm/year in the first year of treatment.11,12 The acceleration of height velocity after GH treatment is more prominent at a higher dosage (50 µg/kg/day) than that at a dosage close to a replacement dosage (25 µg/kg/day).13 These results suggest that children with ISS may not have sufficient endogenous GH secretion to overcome decreased GH sensitivity, therefore, a larger replacement dose of GH results in more increase of the growth response. Studies about the short-term height gains have shown that the change in height SDS can range from none to approximately 0.7 SDS over one year.9 In the present study, GH treatment for 6-month increased yearly height velocity by 6.36 cm/yr, as well as gain in height SDS by 0.57, suggesting that short-term GH treatment in prepubertal children with ISS affects on growth promotion. Moreover, increase in the height velocity and gain in height SDS observed by week 13 revealed stimulatory effect of GH treatment occurring even earlier.
In the present study using GH dosage of 0.37 mg/kg/week (61.7 µg/kg/day), no effect was observed on BA advancement. Previous studies using approximately 30-50 µg/kg/day of GH have also shown that GH does not influence the rate of BA advancement.6,14,15 However, one randomized controlled study using GH with a dosage of 70 µg/kg/day has demonstrated a significant advancement of BA and onset of puberty, suggesting that a higher dosage of GH may accelerate bone maturation and onset of puberty.16 For these reasons, it is suggested that GH dose of 50 µg/kg/day might be closer to the optimum dosage for ISS treatment, and the regulatory authority in the United States has approved GH treatment at doses up to 0.3-0.37 mg/kg/week (50-61.7 µg/kg/day) for children with ISS shorter than -2.25 SDS.17 On the other hand, one study adjusting the GH dosage according to the serum level of IGF-I showed no change in BA advancement and pubertal development even at higher dose (mean GH dose of 110 µg/kg/day), suggesting a large individual variability in this matter.18
Several studies have proved that GH can increase the final adult height in children with ISS. The adult height of children with ISS after GH treatment for 4-7 years was average 3.7-7.5 cm taller than untreated or placebo-treated controls.9,19 Adult height SDS was also significantly greater in the GH treated group (-1.77±0.17 SDS) than in the placebo-treated group (-2.34±0.17 SDS) by 0.57 SDS.9 The effect of GH on adult heights in children with ISS is known to be similar to that of Turner syndrome, or GH deficiency, and similar or slightly less than that in children born SGA.19 The growth response to GH is highly variable, and may be determined by many factors. Some children who developed positive antibodies to GH treatment have found to have no effect on the growth response. It has been proposed that adult height outcome is negatively correlated with age at treatment start and positively correlated with GH dosage, midparental height, height at treatment start, delay in BA, and the first-year response to GH.19,20
Previous studies found that the incidences of adverse events after GH treatment in children with ISS are similar to those in children with other conditions, such as GH deficiency, or Turner syndrome receiving GH therapy.21 One study evaluating the safety of GH in 307 children with ISS showed that 13-14% of GH-treated patients had serious adverse events, which were mainly unrelated to GH exposure, such as hospitalizations for accidental injury or acute illness. Reported adverse events in ISS study were otitis media (8%), scoliosis (3%), hypothyroidism (0.7%), changes in carbohydrate metabolism (0.7%), and hypertension (0.4%). However, GH treatment of children with ISS did not affect the measures of carbohydrate metabolism.22 Other studies have reported no serious adverse events such as peripheral edema, pancreatitis, or pseudo-tumor cerebri.9,19 In the present study, 23 (63.89%) children out of 36 reported 65 adverse events, which seem to be unrelated to GH therapy except two events. Mild pruritus on injection site followed by spontaneous resolution without any intervention was reported twice in a same subject, which was assessed as being related to GH. Moreover, no serious adverse events were reported in this study. All the studies in children with ISS including the present study have demonstrated that GH is safe in treating ISS, at least during treatment. However, GH has a potential influence on carbohydrate metabolism and is possibly associated with tumor formation. As previously reported, mean fasting insulin levels were significantly higher in the GH-treated children with ISS than those in untreated controls.23 However, there are no reports regarding the risk for type 2 diabetes mellitus and metabolic syndrome in adults who received GH treatment for ISS as children, compared to untreated controls.19 The possible associations between GH and malignancy can be explained by the findings that IGF-I regulated by GH has effects on cell proliferation, cell migration, and cell mitosis in vitro. The involvement of IGF system in cancer growth and metastasis has been suggested by many studies that there are abnormal expressions of IGFs in a variety of cancer; that targeted expression of IGF-I in prostate epithelium leads to prostate cancer in transgenic mice, and that high levels of serum IGF-I represent an increased risk of breast, prostate, lung and colon cancer.24 Recently, preliminary report of the French Safety and Appropriateness of Growth hormone treatments in Europe (SAGhE) study demonstrated that mortality rates were increased in adults treated with GH as children. Particularly, those who had received the highest doses showed increased bone tumors or cerebral hemorrhage but not for all cancers.25 However, these results are not yet conclusive and still in debate. The French SAGhE study recommended to secure proper dose (50 µg/kg/day) and continued surveillance through a registry and to perform long-term follow-up studies of GH-treated patients. In addition, there are many ongoing debates about the indications for therapy, psychosocial benefit, improvement of quality of life, and cost issues in the treatment of GH for children with ISS.19
Despite the limiting factors such as small number of subjects, uncontrolled study, and short-term treatment duration, the present study demonstrated the efficacy of GH (Eutropin with a dose of 0.37 mg/kg/week) in children with ISS by showing significant increases in height velocity, height SDS, and serum levels of IGF-I and IGFBP-3 with favorable safety. Nevertheless, further long-term assessment and surveillance for GH-treated children with ISS are warranted.




 XML Download
XML Download