

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
X-linked adrenoleukodystrophy (X-ALD), a fatal neurodegenerative disorder, is caused by a defect in the adenosine triphosphate-binding cassette D1 (ABCD1) gene, which is involved in the peroxisomal oxidation of very long chain fatty acids (VLCFAs).1 Childhood cerebral ALD and adrenomyeloneuropathy (AMN), the two most common phenotypes, account for 70-80% of patients with X-ALD.1,2 Presentation as spinocerebellar degeneration or olivopontocerebellar degeneration have been rarely reported in literature,3,4,5,6,7,8,9 and these reports described a mutation in exon 2 or exon 8 of the ABCD1 gene in these unusual phenotypic variants of X-ALD.4,5,9 Herein, we report a Korean male with X-ALD with an isolated lesion in the cerebellar white matter and dentate nuclei, derived from a de novo mutation in exon 1 at nucleotide position c.277_296dup20 (p.Ala100Cysfs*10) of the ABCD1 gene.
CASE REPORT
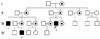
A 37-year-old man (#3 in Fig. 1) is presented with a two-year history of progressive slurred speech, bladder incontinence, ataxia, and memory deterioration. Fortunately, they did not significantly affect his daily function and activities. He had no complicated perinatal history and past medical history, including Addison's disease. He had normal developmental milestones in areas of motor, language, and cognitive skills. On his mother's side, his cousin, who was diagnosed with adult cerebral type ALD at the age of 27, died (#1 in Fig. 1). Also, his nephew was diagnosed with Addison's disease at the age of seven (#2 in Fig. 1). His several aunts and female cousins have also been diagnosed as AMN by only VLCFAs assay.
Upon physical examination, he was alert, cooperative and well nourished. He had no skin rash or pigmentation. His speech was dysarthric without fluency. He had dysmetria in all limbs, difficulties in balancing, and gait ataxia. He reported that he sometimes have diplopia that would last less than 5 seconds. Fundoscopy did not reveal optic atrophy. The rest of the cranial nerve examinations were normal. All limbs were normotonic and normoreflexic. Power in all limbs ranged between medical research council scale, grade 4 and 5. He had normal sensation to pinprick, light touch, and proprioception and skeletal deformities, visceromegaly, or neurocutaneous lesions were not found in this patient. The following laboratory investigations including full blood count, electrolytes, renal function, liver function, venereal disease research laboratory, toxoplasma, rubella, cytomegalovirus and herpes serologies, serum calcium, phosphate, lactate and ammonia, arylsulfatase A, and pituitary hormonal assays were normal. In addition, we could not any abnormal findings of electrophysiologic studies including electrocardiogram, nerve conduction studies, visual evoked potentials, brainstem auditory evoked potentials and somatosensory evoked potentials studies.
Brain magnetic resonance imaging (MRI) taken at the age of 35 year showed notable white matter signal change only on cerebellum and became more apparent after 2 years. There were abnormal signals in the dentate nuclei on the axial and sagittal scans. No contrast enhancement was seen in these lesions with gadolinium injection. The rest of the brain parenchyma was completely normal in signal intensity and morphology without any signal abnormality of visual and auditory pathways. The brain stem and spinal cord were also normal on MRI (Fig. 2). We evaluated the concentration of plasma VLCFAs that it revealed the following (normal range): C22:0 48.29 µmol/L (≤96.3 µmol/L); C24:0 82.28 µmol/L (≤91.4 µmol/L); C26:0 3.09 µmol/L (≤1.3 µmol/L); C24:0/C22:0 ratio 1.70 (≤1.39) and C26:0/C22:0 ratio 0.06 (≤0.023). Phytanic and pristanic acid levels were normal. DNA analysis for ABCD1 gene through direct sequencing showed c.277_296dup20 mutation (p.Ala100Cysfs*10) on exon 1 (Fig. 3). Since the diagnosis, the patient has been taking Lorenzo's oil without any symptom aggravation for about 3 years, even though it is not a proven treatment option.
DISCUSSION
Our X-ALD patient presented progressive slurred speech, bladder incontinence, ataxia and memory deterioration, and had hyperintense lesions in the dentate nuclei of cerebellum on MRI. Our patient's symptoms were very similar to the clinical manifestations of spinocerebellar ataxia, especially in the absence of adrenal insufficiency. However, brain MRI did not show any lesions in the corticospinal tract in our patient. Moreover, he was suspected to have X-ALD because of the family history. The diagnosis of X-ALD was screened by high levels of VLCFAs in the plasma and could be confirmed by gene study.
X-ALD shows a wide range of phenotypic expression and is subdivided into various categories: childhood cerebral, AMN, adolescent or adult cerebral, Addison's disease only, olivo-ponto-cerebellar forms.10 AMN patients generally have spinal cord dysfunction, which leads to the initial symptoms that include difficulties in walking or a change in the walking pattern and eventually presents sphincter disturbance, sensory changes and pain due to involvement of long tracts of the spinal cord and peripheral nerves. Approximately 30-40% of patients involve white matter in cerebrum in AMN. In the rare form of X-ALD, patients presented mainly cerebellar and brainstem involvement in adolescence or adulthood.3,11,12,13 To the best of our knowledge, there are reports of 17 cases of adrenoleukodystrophy presenting as spinocerebellar degeneration or olivopontocerebellar atrophy worldwide. Clinical presentations of these cases were similar to spinocerebellar ataxia or olivopontocerebellar atrophy similar to our patient.3,4,5,6,7,8,9,11,12,13,14
White matter T2 signal hyperintensity lesions involving parieto-occipital area has been reported as the most common MRI abnormalities in childhood cerebral X-ALD. The involvement of frontopontine or corticospinal projection fibers and cerebellar white matter at presentation are more frequent in the adult group. It is known that cerebellar lesions accompany the other lesions, such as cerebral white matter or deep brain structures in usual cerebellar X-ALD.15,16,17 However, brain MRI in our patient showed abnormal signals only in the cerebellar white matter and dentate nuclei, unlike the previously reported cases.8,12,15,16,17
No reports has described a mutation of exon 1 of the ABCD1 gene in spinocerebellar variant of X-ALD. In our patient, we have found a de novo mutation in exon 1 of the ABCD1 gene in this family. This is the first report of a c.277_296dup20 mutation (p.Ala100Cysfs*10) on exon 1 of the ABCD1 gene causing the cerebellar variant of X-ALD. A duplication of 20 base pair at nucleotide position c.277_296 was found. c.277_296dup20 mutation causes a substitution of the 100th amino acid from alanine to cysteine and finally 109th codon is changed to stop codon, p.Ala100Cysfs* 10. It might cause a dysfunction of adenosine triphosphate-binding cassette transporter by newly formed stop codon by duplication. So far, 1251 mutations have been reported in the ABCD1 gene, 587 (46.9%) of which are unique mutations, as listed in the X-ALD database (http://www.x-ald.nl). In this respect, there seems to be no direct correlation between genotype and clinical phenotype and other genetic or environmental factors can influence to the phenotype variance of X-ALD.
In summary, we report an unusual presentation of X-ALD only with an isolated lesion of the cerebellar white matter and dentate nuclei. It is strongly advised to consider X-ALD as a differential diagnosis in patients with isolated cerebellar degeneration symptoms.


 XML Download
XML Download