

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cholesterol is an essential component of the plasma membranes of eukaryotic cells and plays important roles in membrane fluidity, permeability, receptor function, and ion channel activity.1-4 The concentration of cholesterol is focally high in the submicroscopic areas enriched with sphingolipids and gangliosides. These microdomains, known as lipid rafts, are associated with the regulation of transmembrane receptors, especially tyrosine kinase receptors and G protein-coupled receptors. If cholesterol in lipid rafts is depleted by treating cells with methyl-β-cyclodextrin (MβCD), the activities of receptors are altered in a cell-specific and/or receptor-specific manner.5,6
Mucin hypersecretion results in inflammatory airway diseases such as rhinitis, sinusitis, and bronchitis. Various genes and cytokines are related to mucin secretion. Among human mucin genes, MUC5AC is recognized as the major airway mucin gene in the airway epithelium.7-9 Recently, it has been determined that IL-1β, a well-known proinflammatory cytokine, induces MUC5AC gene over-expression in NCI-H292 cells via extracellular signal regulated kinase (ERK)/p38 mitogen-activated protein kinase (MAPK) cascade.10,11 As MAPK signal transduction is associated with regulation of MUC5AC, it has been postulated that cholesterol depletion can influence MUC5AC gene expression in NCI-H292 cells by changing the activity of MAPK. Other studies demonstrated that MAPK activation was regulated by MβCD treatment in keratinocyte, NIH3T3 cells, and cultured human dermal fibroblasts.12-14 However, the relationship between MAPK and MUC5AC in human airway epithelial cells is still unknown. Therefore, we sought to investigate the effects of cholesterol depletion on the expression of MUC5AC in NCI-H292 cells. If the expression of MUC5AC is related to cholesterol depletion in the cell membrane of airway epithelial cells, we also sought to investigate whether the alteration of MUC5AC expression is related to MAPK activity.
MATERIALS AND METHODS
Cell culture
Human pulmonary mucoepidermoid carcinoma cell lines (NCI-H292 cells) from American Type Culture Collection (Rockville, MD, USA) were cultured respectively in RPMI 1640 (Gibco BRL, Grand Island, NY, USA) and DMEM (Gibco BRL, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Cellgro, Hemdon, VA, USA) in the presence of 2 mM L-glutamine, penicillin (100 µg/mL) and streptomycin (100 µg/mL) at 37℃ in a humidified chamber with 95% air and 5% CO2. When cultures reached 60-80% confluence, the cells were incubated in each medium containing 0.5% fetal bovine serum for 24 hours, after which they were rinsed with phosphate buffered saline (PBS) and exposed to the indicated concentrations of reagents sub-sequent human recombinant IL-1β (R&D Systems, Minneapolis, MN, USA) treatment. Some cultures were pretreated with MβCD (Sigma Chemical Co., St. Louis, MO, USA) for 1 hour before being exposed to IL-1β. IL-1β was dissolved with PBS containing 0.1% bovine serum albumin.
Materials
MβCD was purchased from Sigma Chemical Co.; IL-1β was purchased from R&D Systems; anti-phospho-Type I IL-1 receptor (phospho-Y496) antibody (IL-1RI) was purchased from Abcam Co. (Cambridge, MA, USA); anti-phospho-p44/42 MAPK (Thr202/Tyr204) antibodies and anti-phospho-p38 MAPK (Thr180/Tyr182) antibodies, were purch-ased from Cell Signaling Co. (Beverly, MA, USA).
Determination of cell viability (MTS assay)
Cell viability was determined by MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2(4-sulfophenyl)-2H-tetrazolium, inner salt] and electron coupling reagent (phenazine ethosulfate) assays. The cells were seeded on 96 well plates at 2000 cells/well. After serum starvation for 24 hours, cells were then treated MβCD with at various concentrations (0, 0.5, 1, 2%) for 1 hour. After the exposure period, the media were removed. Thereafter, the medium was changed and incubated with reagent (CellTiter96 AQueous One Solution Proliferation Assay: Promega, Madison, WI, USA) for 1 hour at 37℃, 5% CO2. Optical density was measured by spectrophotometer at 492 nm.
Measurement of intra-membranous cholesterol
Cells were rinsed twice with 1 mL cold PBS, and lipids were extracted with chloroform-methanol 2 : 1 (v/v). Homogenized cell lysates were centrifuged for 10 min at 14000 rpm. The organic phase was transferred to a clean tube, dried under vacuum, and re-dissolved in 20 µL 2-propanol and 10% Triton X-100. One µL per assay was used, and it was adjusted to 50 µL with cholesterol reaction buffer in the wells of a 96-well plate. Cholesterol levels were measured using a modified microenzymatic fluorescence assay (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer's protocol. Samples were incubated at 37℃ for one hour. A spectrofluorometer (excitation 544 nm and emission 590 nm) was used for measurement. The protein concentrations of the supernatant were measured by bicinchronic acid protein assay using bovine serum albumin. The results were described as the ratio of cholesterol/cell protein (µg/mg).
Filipin staining
Cells, cultured in the polysin-coated cover slip, were divided into two groups; a control group and an experimental group, and treated with MβCD. Subsequently, cells were rinsed with cold PBS and fixed on ice using 4% paraformaldehyde. The cells were rinsed again with cold PBS for 10 min, and stained at room temperature with 100 µg/mL of filipin (Sigma Chemical Co., St. Louis, MO, USA) for 2 hours. After being rinsed once with PBS, the cells were observed through a fluorescence microscope with a UV filter set (340-380 nm excitation, 40 diachronic, 430 nm long pass filter).
Reverse transcription-polymerase chain reaction (RT-PCR) analysis of MUC5AC mRNA
The total RNA was isolated from cells under each condition using TRI-reagent (Molecular Research Center, Cincinnati, OH, USA). Total RNA (1 µg/20 µL) was reverse transcribed into cDNA using random hexanucleotide primers and Moloney murine leukemia virus reverse transcriptase (Gibco-BRL), and MUC5AC cDNA was amplified by PCR using a Perkin-Elmer Cetus DNA Thermal Cycler (Perkin-Elmer, Norwalk, CT, USA) using the previously described method.11 β2-microglobulin (β2M) was used as a control gene for RT-PCR.
Real-time polymerase chain reaction of MUC5AC mRNA
Primers and probes were designed using Applied Biosystems Primer Express software and purchased from Applied Biosystems (Carlsbad, CA, USA). Commercial reagents (TaqMan Universal PCR Master Mix; Applied Biosystems) and conditions were applied in accordance with the manufacturer's protocol. One µg of cDNA (reverse transcription mixture) and oligonucleotides with final concentrations of 800 nM for primers and 200 nM for TaqMan hybridization probes were analyzed in a 25 µL volume using a previously described method.11 Relative quantities of MUC5AC mRNA were obtained using a comparative cycle threshold method and normalized using β2M as an endogenous control. Expression was reported as a ratio of MUC5AC intensity to β2M intensity.
Western blot analysis
Cell lysates were made into cultured cells using a radioimmunoprecipitation assay buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS). Protein quantities were measured via bicinchronic acid protein assay and bovine serum albumin was also added. Next, the protein was placed into lanes by 30 µg and electrophoresis was performed. We respectively used 4-12% gel (Invitrogen, Grand Island, NY, USA) in IL-1RI analysis and 8% SDS-polyacrylamide gel in MAPK analysis for electrophoresis. Afterwards, they were transferred to the nitrocellulose membrane. We then performed Western blot analysis using a previously described method.11 We used p-IL-1RI (1 : 500), p-ERK (1 : 1000), and p-p38 MAPK (1 : 1000) antibodies.
Statistics
The experiment was performed at least three times and the mean value and the standard deviation for all experiments were calculated. The repeated measures ANOVA test was used to analyze differences, and multiple comparisons were added. Cases in which the p-value was less than 0.05 were defined as significant.
RESULTS
Cell viability by cholesterol depletion of cell membranes in NCI-H292 cells
We treated cells with MβCD at various concentrations (0, 0.5%, 1%, 2%) for 1 hour, and examined cell viability. 0.5% MβCD showed 88±8% cell viability compared to the control group (no treatment with MβCD) and 1% MβCD showed 83±10% and 2% MβCD showed 60±5% (Fig. 1). Cells exposed to 1% MβCD exhibited normal viability, but those exposed to 2% MβCD showed loss of cell viability. Therefore, we used 1% MβCD in the following experiments.
Verification of cholesterol depletion by MβCD treatment
To verify whether intra-membranous cholesterol was depleted after 1 hour of treatment with 1% MβCD, intra-membranous cholesterol level analysis and filipin staining were performed. After treatment with 1% MβCD for 1 hour, the intra-membranous cholesterol/protein ratio was calculated and the data for the experimental group was compared to that of the control group, which had not been treated with MβCD. As a result, the relative ratio of intra-membrane cholesterol/protein was 0.17±0.07. This result indicates that MβCD significantly decreased the cholesterol level in the cell membranes (p<0.05) (Fig. 2A).
With regards to the filipin staining, in the control group, filipin was observed to adhere specifically to cholesterol along the cell membrane. However, in the MβCD-treatment group, the intensity of fluorescence decreased significantly (Fig. 2B).
Suppression of MUC5AC expression after treatment with MβCD
We evaluated whether MβCD suppressed IL-1β-induced MUC5AC gene expression using RT-PCR and real time-PCR, as well as whether this suppression occurred IL-1-receptor-specifically. Cells were pretreated with 1% MβCD for 1 hour, after which the media was freshly changed and cells were incubated with IL-1β (10 ng/mL) for 24 hours. We then picked MUC5AC mRNA and examined its expression through RT-PCR and real-time PCR. The expression of p-IL-1RI was examined by Western analysis. We found that IL-1β induced the expression of MUC5AC, but this increase was significantly decreased by treatment with MβCD in RT-PCR and real-time PCR analysis (control : MβCD : IL-1β : IL-1β+MβCD=1 : 0.9±0.2 : 5.3±0.8 : 0.8±0.3) (p<0.05) (Fig. 3A and B). The expression of p-IL-1RI increased according to the IL-1β treatment, but MβCD treatment significantly decreased the expression of p-IL-1RI (Fig. 3C).
MAPK's changes by MβCD treatment
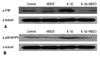
To figure out whether the decrease in MUC5AC expression induced by MβCD treatment was dependent upon the depression of MAPK activity, we pre-treated cells for 1 hour with 1% MβCD. Then the media was freshly changed and cells were incubated with IL-1β for 20 min. A no MβCD or IL-1β treatment group was used as a negative control group and only IL-1β was administered in the positive control group. After each experiment, protein was obtained and a Western blot analysis of p-ERK and p-p38 MAPK was performed. Elevated expression of p-ERK in only IL-1β-treated cells decreased markedly in both IL-1β and MβCD-treated cells (Fig. 4A). Expression of p-p38 was also reduced to the negative control level in both IL-1β and MβCD-treated cells (Fig. 4B).
DISCUSSION
MβCD is a compound that specifically combines with cholesterol, deletes it, and then disrupts the lipid raft. Due to this effect, MβCD affects kinases of plasma membrane receptors and may cause alterations to cell proliferation.15,16 These facts mean that MβCD induces inhibition of cell proliferation according to cell type and concentration of MβCD. It is thus necessary to determine the appropriate concentration of MβCD. Our results regarding cell viability were above 80% when using 1% MβCD; therefore, this concentration was considered safe for maintaining cell survival. Although we found the appropriate concentration of MβCD, we fixed the treatment time at one hour. In other studies, the duration of treatment was 1 or 2 hours and, after reviewing previous results, we fixed our duration as 1 hour.17-19 In the preceding experiments, we treated cells for 2 hours at a 1% concentration, but cell proliferation was remarkably inhibited (data not shown), and we decided to use one hour as the experimental time.
Our next step was to verify whether cholesterol was depleted by MβCD in NCI-H292 cells via intra-membranous cholesterol level analysis and filipin staining. Through the intra-membranous cholesterol analysis, we found that MβCD depleted the cholesterol of the plasma membrane. However, cholesterol is distributed both intramembranously and intracellularly, and intra-membranous cholesterol level analysis examines the relative ratio of cholesterol/protein.1,20,21 Therefore, we determined whether the cholesterol of the cell membrane was depleted through filipin staining. By staining filipin that selectively combines with cholesterol, we verified that filipin on the membrane was further decreased in MβCD-treated cell lines as compared to the control group. From this result, we confirmed that MβCD selectively depletes the cell membrane's cholesterol.
Because MUC5AC expression is induced by IL-1β in NCI-H292 cells, we applied MβCD before IL-1β treatment.11 According to our results, MUC5AC expression was significantly decreased. Next, we wondered whether these results were induced by the disruption of a ligand-specific receptor such as IL-1 receptor or not. Therefore, we examined whether the phosphorylation of IL-1 receptor is induced by IL-1β and whether such activation of the ligand-specific receptor is decreased by MβCD. As a result, the expression of p-IL-1RI was increased by IL-1β treatment, but this increase was significantly decreased by MβCD treatment. Thus, we discerned the suppression of MUC5AC expression by MβCD to be an IL-1 receptor-specific reaction.
In order to reveal the possible signal pathway by which MUC5AC is decreased by MβCD, we examined the MAPK cascade. In our previous study of NCI-H292 cells, IL-1β maximally induced activation of p-ERK and p-p38 in the 20 min after treatment.11 Thus, our experiments were performed 20 min after IL-1β treatment. We found ERK and p38 MAPK to be associated with a decrease in MUC5AC expression by MβCD treatment. Previous studies have investigated the relationship between MAPK and cholesterol depletion. Jans, et al.21 found that cholesterol depletion of the cell membrane in keratinocyte induced prolonged phosphorylation of p38 MAPK. Kim, et al.14 reported that cholesterol depletion brought about activation of ERK and JNK in human dermal fibroblasts. Calleros, et al.13 showed that low cell cholesterol levels increased NFκB activity by increasing p38 activity. The difference of MAPK activation between our study and others seems to be related to alterations in respective receptors. MβCD treatment triggers MAPK cascade, and changes lipid raft in keratinocyte, NIH3T3 cells, and cultured human dermal fibroblasts.12-14 Thereafter, the activation of MAPK is followed in a cell-specific manner. On the contrary, NCI-H292 cells express a very low level of MAPK in unstimulated state. The cells stimulated with IL-1β show MAPK activation.11 Disintegration of lift raft leads to alterations in IL-1β receptor, and the activation of MAPK is attenuated even though IL-1β is challenged. This phenomenon is also found in human promonocytic THP-1 cells. If THP-1 cells are challenged with lipopolysaccharide, MAPK such as ERK 1/2 is activated. With MβCD pre-treatment, this MAPK activation is attenuated in spite of lipopolysaccharide treatment.22 These results coincide with our results. Modulation of lipid raft may represent a mechanism for regulation of MAPK activaton. In addition, two studies reported that EGFR ligands induce MUC5AC production through EGFR signaling kinase cascade in the airway system, and are also related to MAPK activation.23,24 Thus, further studies on IL-1β receptor and EGFR signaling cascades are needed.
Mucin is hypersecreted in many respiratory diseases such as rhinitis, sinusitis, nasal allergy, and chronic bronchitis.10 Hypersecretion of mucin results in clinical problems such as rhinorrhea, nasal stuffiness, and sputum. Accordingly, regulation of MUC5AC can be a new strategy for treating respiratory diseases because it decreases mucus hypersecretion. Furthermore, IL-1 receptor was recently considered a novel potential therapeutic target for bronchial asthma.25
In this study, we showed that MUC5AC expression in NCI-H292 cells is significantly decreased by treatment with MβCD, which depletes cholesterol in the cell membrane. We also found that the decrease of MUC5AC expression is dependent upon the activation of ERK and p38 MAPK. In conclusion, cholesterol depletion in the cell membrane may be considered as anti-hypersecretory method because they effectively inhibit mucus secretion in human airway epithelial cells.



 XML Download
XML Download