

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE) myopathy, otherwise known as Nonaka myopathy1 or hereditary inclusion body myopathy2,3 is an autosomal recessive neuromuscular disorder characterized by early adult-onset weakness of the distal muscles of the lower limbs. The tibialis anterior is typically affected, while the quadriceps group is typically spared.3 Patients with GNE myopathy exhibit mildly elevated serum creatine kinase (CK) levels.
This disease is caused by mutations in the UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase (GNE) gene that regulates the synthesis of sialic acid.4,5 The GNE gene encodes a bifunctional protein with two enzymatic activities: UDP-GlcNAc2-epimerase (GNE) and ManNAc kinase.6 More than 70 GNE mutations have been described to be associated with GNE myopathy in patients of different ethnic origin. Two mutations are frequently observed in two specific ethnic groups, M712T in Middle Eastern Jews and V572L in Japanese patients, suggesting a founder effect.2,7,8
Although the molecular mechanism by which the mutations in the GNE gene cause the muscle degeneration seen in GNE myopathy remains unclear, it has been proposed that the mutations may lead to defective sialylation of muscle tissue.9,10 One characteristic histopathological feature is the presence of numerous rimmed vacuoles, which are immunoreactive to various proteins.11 Despite these findings, the pathophysiological mechanisms underlying rimmed vacuole formation remain to be elucidated.
The clinical spectrum of GNE myopathy varies, as the prominent initial symptoms and advanced symptoms are different for each patient.7,12 However, it is not clear how the same GNE gene mutations can result in different phenotypes. Herein, we report clinical characteristics and molecular genetic analysis of Korean patients with GNE myopathy.
MATERIALS AND METHODS
Subjects
Twenty-one GNE myopathy patients were included in this study, conducted from 2004 to 2011. All patients exhibited the following suspicious features of GNE myopathy: 1) autosomal recessive or sporadic; 2) early adult onset; 3) the initial symptom of gait disturbance due to weakness, typically in the anterior compartment of the distal leg; and 4) myopathic changes on electromyography. All patients were evaluated neurologically and were referred for genetic testing with informed consent. Of those, only patients with confirmed mutation of the GNE gene by DNA analysis were enrolled in this study.
Clinical data collection
Clinical data for each patient were obtained by reviewing all records available in the electronic medical records at the hospital. Such data included the patients' gender, age at symptom onset, family history, clinical history, serum CK level, neurologic examination, findings of muscle biopsy, muscle imaging findings and electrophysiologic features.
Genetic analysis
After informed consent was obtained, blood samples were obtained from suspicious patients and DNA study was performed. Mutation of the GNE gene (9p13.3) was confirmed by DNA direct sequencing analysis in all patients. Genomic DNA was extracted from peripheral blood leukocytes using a genomic DNA extract kit (Easy-DNA kit, Invitrogen).
All 11 coding exons (exon 2-12) of the GNE gene were amplified by performing PCR using forward and reverse primers. Mutations were identified with reference to the Human Gene Mutation Database.
RESULTS
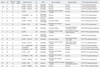
Clinical and pathological data, as well as data on GNE mutations, of the Korean patients with GNE myopathy are summarized in Table 1. The mean onset age was 23.8±8.8 years (mean±SD). Among these patients, twelve patients were female and nine patients were male. The mean onset age in females was 23.6±9.9 years (mean±SD), and the mean onset age in males was 24.1±7.6 years (mean±SD). Patient serum creatine kinase levels were slightly to moderately elevated, ranging from 41 to 2610 IU. Four patients presented with a family history of GNE gene mutation. Except for eight patients, all of the patients presented initially with only distal muscle weakness in the lower extremities. However, these patients showed different sites of progressing weakness. Two patients initially showed weakness in the upper extremities, while the others initially showed proximal or both proximal and distal weakness in the lower extremities. Electromyogram studies showed a myopathic or mixed pattern in all patients. Eleven of twenty-one patients underwent muscle biopsy. On muscle biopsy, five patients exhibited rimmed vacuoles of myofibers. Five patients underwent muscle imaging. Except for one patient, the tibialis anterior was the most severely affected muscle, while the quadriceps were relatively spared. Direct nucleotide sequencing disclosed fifteen compound heterozygous GNE mutations and six homozygous GNE mutations. In addition, one patient was found to have only one heterozygous mutation. Because single heterozygous mutations was not confirmed to cause GNE myopathy, this patient was excluded from this study. Of these twelve different mutations, D208N and His157fs were novel. The others have been previously reported.13 The most common mutation was V572L, followed by C13S.
DISCUSSION
This study comprised the largest series of Korean patients with GNE myopathy. As previous study revealed only genetic mutations,13 this study attempted to document clinical, pathological and genetic characteristics of Korean patients with GNE myopathy. Here, we examined the relationship between genotype and clinical phenotype. The clinical course of our patients varied, as seen in Table 1. The initial site of weakness and the progressing site of weakness were different for each patient. Among twenty-one patients, thirteen patients showed typical GNE myopathy phenotype.2 Their mean age of onset was around 25 years and their initial symptom involved severe anterior tibialis muscle weakness, relatively sparing the quadriceps muscles and upper extremities. Patients of GNE myopathy have been described to have high CK levels.4,14 In the present study, however, serum CK was slightly to moderately elevated or within normal limits in seventeen patients, and only four patients showed high CK levels (more than 1000 IU).
UDP-N-acetylglucosamine 2-epimerase/N-acetylmannosamine kinase, a rate-limiting enzyme that catalyzes the biosynthesis of sialic acid, has two functional domains that work independently, an epimerase domain and a kinase domain.8 Various GNE mutations have been identified in GNE myopathy patients of various ethnic origins. The mutation M712T is the most common in Jewish hereditary inclusion body myopathy. And the most frequent mutation in Japanese GNE myopathy patients is the V572L mutation.2,7 Since the V572L mutation was also shown to be the most frequent mutation in Koreans,13 a common founder effect might exist between these populations. The most common mutation in China is L508S,15 and V696M in Thailand.16
Several articles have suggested a relationship between the clinical features and site of mutation. The typical clinical features of GNE myopathy result from homozygous mutations in the kinase domain, whereas involvement of the quadriceps muscles results from compound heterozygous mutation in both the epimerase and kinase domain.2 However, some patients (from the same family) displayed the same compound heterozygous mutations in different domains of the protein, but demonstrated different phenotypes.17 Even with the same homozygous mutation, there appears to be heterogeneities in the severity of the clinical presentations.7,14 Also, our data showed that there was no relationship between clinical features and mutation site. Therefore, we suggest that neither homozygous nor compound heterozygous models are correlated with disease phenotype or disease severity. Recently, V572L mutation was suggested to be associated with disease severity.18 However, for more detailed information on the genotype-phenotype correlation and the effect of the mutation site, further clinical and genetic analyses are needed, along with review of the natural course of this disease.
 XML Download
XML Download