

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Long QT syndrome (LQTs) is an uncommon disease causing sudden cardiac death,1 and its first line treatment has been known to be β-blocker. But we experienced a patient with atypical clinical presentation, which was aggravated by β-blocker and alleviated by mexiletine. We also report that epinephrine provocation test can play an important role in diagnosis and monitoring of drug efficiency in LQTs.
CASE REPORT
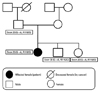
A 15-year-old female middle school student was referred to our hospital because of multiple syncopal episodes. She had experienced four syncopal episodes at school during the last three years. All of these episodes occurred while resting or in class. Her mother noticed the patient's very unusual snoring sound with transient loss of consciousness twice at night, and what seemed like agonal respiration. After that, she had palpitations with presyncope while writing in class. She had a past history of patch repair surgery for ventricular septal defect (VSD) at the age of 5 months old, however, there was no abnormality in the physical examination or laboratory findings, and echocardiography showed her heart to be completely normal morphologically and functionally. The only abnormal finding was prolonged QTc (629 ms) in surface 12 lead electrocardiography (ECG) (Fig. 1A). She had no family history of sudden death or other genetic heart disease (Fig. 2). At the time when she was referred to our hospital, she was taking low dose of propranolol. Because ECG at the time of presyncope or syncope had not been documented and her clinical presentation was unusual, we stopped propranolol and performed an epinephrine test.
Epinephrine provocation test
After skipping propranolol for 5 times its half life, an epinephrine provocation test was performed and mean QTc was measured after epinephrine test.2,3 Mean QTc was significantly prolonged (637.0-48.6 ms; between 585 and 646 ms), and frequent single or couplets of ventricular premature beats appeared (Fig. 3A). We then repeated the epinephrine test with β-blocker medication. With propranolol 120 mg per day, mean QTc was prolonged as 628.5±62.7 ms (between 618 and 634 ms), and non-sustained polymorphic ventricular tachycardia (VT; 200 bpm) appeared following R-on-T phenomenon and spontaneously ended within 8 sec without syncopal event (Fig. 3B). There were two different premature ventricular contraction (PVC) morphologies, and non-sustained VT initiating PVC was localized by 12 lead ECG (Fig. 1B). The origin of PVC seems to be left ventricular (LV) high septum just below the (left) distal HIS bundle. QRS duration is relatively narrow and purely negative in lead aVR and aVF, but initially positive in all other leads, suggesting LV peri-Hissian PVC related to membranous VSD repair. After increasing the dosage of propranolol to 160 mg per day for 3 days, sustained polymorphic VT was induced spontaneously, requiring external defibrillation, and syncope and tonic clonic seizure were documented during the epinephrine provocation test (Fig. 3C). Mean QTc was prolonged as 644.0±33.7 ms (between 619 and 668 ms). However, after stopping propranolol for 4 days, no arrhythmic event was reproducible during the epinephrine provocation test (Fig. 3D).
Implantable cardioverter defibrillator (ICD)
Because the patient's arrhythmia could not be controlled by β-blocker, she underwent ICD implantation. She returned to the emergency room due to ICD shock in the 3rd month after ICD implantation. ICD EGM documented sustained polymorphic VT with cycle length 220 ms and successful ICD therapy. Therefore, we repeated the epinephrine provocation test while medicating with mexiletine 600 mg per day, and there was no event except for QTc lengthening >30 ms (Fig. 3E). She was discharged with mexiletin 600 mg per day and an increase in atrial pacing rate to higher than 70 bpm. During mexiletine medication, there was no VT episode in ICD interrogation for 3 months. However, she arbitrarily stopped the anti-arrhythmic drug due to weakness, and received appropriate ICD shock 8 months after stopping the medication. Therefore, she is presently taking mexiletine 600 mg a day.
DNA isolation and genetic analysis
We genotyped 155 loci from exon 2 to 28 of the SCN5A gene,4 and G>A, R1192Q (rs41261344; reference sequence: NM000335) was identified at the exon 20 sequence. However, same base pair change was found in SCN5A genetic analyses in her father and siblings who did not show the phenotypes of LQTs (ECG or symptom) (Fig. 2). Therefore, it seems to be genetic polymorphism in SCN5A, but not a mutation.
DISCUSSION
Over the past few decades, β-blocker has remained as the most effective medication for LQTs, functioning by shortening the repolarization period and reducing the duration of ventricular tachyarrhythmia episodes.5,6 However, the drug failure rate of β-blocker is significantly higher in patients with LQTs type 3,7 and mortality and morbidity are also higher in LQTs type 3 than those with types 1 or 2. LQTs type 3 accounts for about 5-10% of LQTs.8 Nevertheless, β-blocker has never been reported to aggravate Torsades de Point (TdP) in patients with LQTs type 3. Genetically, LQTs type 3 has been known to be due to the mutation of SCN5A domain which results in a "gain of function in sodium channel" and prolongs action potential duration and QTc. Consequently, the pharmacological blocker of sodium current, mexiletine, shortens the QTc interval and rescues the defect of the SCN5A mutation.9,10-12 However, it is not clear whether gene-specific medication, such as mexiletine, is able to prevent sudden cardiac death in patients with LQTs type 3. The SCN5A gene, consisting of 28 exons and 2016 amino acids, is highly expressed in the human myocardium.13 The missense, a splice-donor, or frame-shift mutations in SCN5A coding regions have been known to be associated with Brugada syndrome, sudden infant death syndrome, and Lev-Lenegre disease.14
In our patient, the clinical presentation of LQTs was unusual (agonal respiration and syncope at rest) and the epinephrine test played a remarkably important role in identifyinggenetic subtype of LQTs and determining an appropriate treatment. First line drug treatment with β-blocker aggravated TdP, and mexiletine suppressed arrhythmic event in this patient. Without the epinephrine provocation test, β-blocker might have been prescribed and harmed this patient with life-threatening channelopathy. Although Vyas, et al.15 reported low predictive value of epinephrine test in LQTs type 3, Shimizu, et al.3 described that it still has an important diagnostic value in LQTs type 3. Ethnic difference or undiscovered genetic polymorphism of SCN5A may play some roles in this difference. Therefore, the epinephrine test can be an important diagnostic and drug monitoring test, and complementary to genetic diagnosis.
R1193Q has been implicated in both LQTs type 3, a gain of function disease,16 and in sudden unexplained nocturnal death syndrome, a loss of function disease.17 At the same time, R1193Q is considered as a polymorphism in Asians with an allele frequency of 8%.4,18 R1192Q in the present case is not distinct from R1193Q in the original hH1 sequence. Therefore, additional polymorphisms in SCN5A may contribute to R1193Q-related QTc prolongation, and we cannot exclude overlap phenotype of LQTs type 3 with Brugada syndrome.19
In conclusion, we experienced a patient with clinically type 3 LQTs who was aggravated by β-blocker and alleviated by mexiletine. The epinephrine provocation test played a remarkably important role in the diagnosis and monitoring of drug efficiency in this patient.


 XML Download
XML Download