

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pancreatic neuroendocrine tumors (PNETs), one of the rarest neoplasms, occur in fewer than one in 100000 people per year and represent 1-2% of all pancreatic tumors.1 PNETs arise in all ages with a peak incidence between 30 and 60 years.1 Their incidence is thought to be increasing over the past 20 years.2 During the 1980s and 1990s, the term "carcinoid" was used, but this term was confusing for pathologists and clinicians. Since 2000, the terms "neuroendocrine tumor" and "neuroendocrine carcinoma" have been introduced to describe neuroendocrine tumors of the gastroentero-pancreatic system.3
PNETs can be classified as functional or non-functional tumors based on symptoms and endocrinologic laboratory tests.4 Functional PNETs secrete biologically active peptides, such as insulin, gastrin, glucagon, somatostatin, and vasoactive intestinal polypeptides.4 Most functional tumors cause glycemic symptoms, such as hypoglycemia, but most non-functional tumors are found by chance. PNETs can be sporadic or may be part of genetic syndrome, such as multiple endocrine neoplasia type 1 syndrome, von Hippel-Lindau disease, neurofibromatosis type 1, and tuberous sclerosis.3
Approximately 60% of patients with PNET have been reported to have liver metastasis at presentation. However, the slow growth pattern of PNET along with improvement in the methods of pre- and intra-operative tumor localization, more aggressive treatments, including surgical interventions, have lead to better outcomes.3 Even though patients with PNET often have liver metastasis, 5-year survival can exceed 80% with liver resection or resection of the primary tumor and multimodal medical therapy.5 On the other hand, a much poorer 5-year survival of 29% has been reported among groups in which primary tumors were not resected.6
There have been some reports concerning the prognostic factors for predicting survival and disease progression of PNETs. Functional status, primary mass size, resectability, lymph node metastasis, distant metastasis, and location of tumor have been reported as prognostic factors.7-9 Up to now, prognostic factors have been inconclusive, because there are no consistent prognostic factors.
The aim of our study was to evaluate the prognostic factors for predicting survival and disease progression in PNETs.
MATERIALS AND METHODS
Patients
Thirty seven patients diagnosed with PNET at Severance Hospital, Yonsei University, in Seoul, South Korea, between November 2005 and March 2010, were enrolled in this study. The clinical and laboratory data of patients were obtained from a retrospectively enrolled database of the patients. As candidate predictive factors for survival or disease progression, clinicopathological parameters, including patient gender, age, tumor size, location, endocrine function, duct invasion, resection of primary tumor, distant metastasis, lymph node metastasis, were investigated from a database of the enrolled patients. Diagnosis of PNET was confirmed by pathologists via immunohistochemical staining (chromogranin A, synaptophysin), of a surgical specimen or biopsy sample.
Data analysis and statistical considerations
The primary end points were overall survival (OS) and progression-free survival (PFS). OS was calculated from the date of diagnosis until death from any cause or the patient's last visit to the hospital. PFS in cases of resected tumors was calculated from the date of operation until the date of recurrence or the day of the last radiological evaluation (computed tomography or magnetic resonance imaging). PFS in cases of unresected tumors was calculated from the date of diagnosis until the date of radiological evaluation, demonstration of tumor size increase, or the day of the last radiological evaluation.
For univariate analysis, OS and PFS were calculated using Kaplan-Meier methods. For multivariate analysis, OS and PFS were calculated using the Cox regression method with a 95% confidence interval. All analyses were performed with the SPSS statistical program (version 18.0; SPSS Inc., Seoul, Korea). A p-value of less than 0.05 was considered statistically
significant.
RESULTS
Baseline characteristics of patients
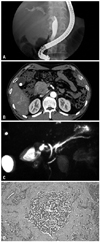
The baseline demographic and clinical characteristics are summarized in Table 1. The patient population included 17 men and 20 women, and the mean age of the patients was 50.0±15.0 years. The most common clinical symptom was hypoglycemia (62.5%) in functioning tumors, but in non-functioning tumors, 69.0% had no specific symptoms. Tumor size ranged from 6 to 100 mm (average 28.08±19.29). Eight cases (21.6%) were listed as functioning tumors and 29 cases (78.4%) as non-functioning tumors. Five patients (14%) had bile duct or pancreatic duct invasion, confirmed by imaging study (4 patients) or pathologic findings (1 patient) (Fig. 1). A total of 13 patients (35.1%) had distant metastasis. Among them, 10 patients (27%) had liver metastasis. Twelve patients (32%) were positive for lymph node metastasis. There were 25 patients (68%) with tumors located at the head of the pancreas. According to American Joint Committee on Cancer/Union for International Cancer Control (AJCC/UICC) staging, there were 21 (56.8%), 6 (16.2%), 0 (0%), 10 (27.0%) cases of stages I, II, III, and IV tumors, respectively. According to European Neuroendocrine Tumors Society (ENETS) staging, there were 16 (43.2%), 6 (16.2%), 5 (13.5%), 10 (27.0%) cases of stages I, II, III, and IV tumors. The mean follow-up period was 23.3±16.6 months (Table 1).
Prognostic factors related to overall survival (OS)
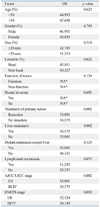
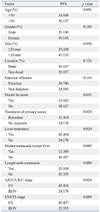
As of March 2010, 5 patients (13.5%) died. The cumulative survival rate was 94.6% at 1 year and 91.9% at 2 years. Univariate analysis of clinical factors showed that primary tumor without resection (p=0.002), with liver metastasis (p=0.002), or AJCC/UICC stage III/IV (p=0.002) were more likely to demonstrate shorter OS (Table 2). Without liver metastasis, the 5-year survival rate was 96.3%, but it was 60% for the group with liver metastasis (Fig. 2A). If the primary tumors were resected, the 5-year survival rate was 96.3%, where as it was 60% for the group in which primary tumors were not resected (Fig. 2B).
Prognostic factors related to progression-free survival (PFS)
As of March 2010, 9 patients (24.3%) showed disease progression. The cumulative progression free survival was 89.2% at 1 year and 81.1% at 2 years. Median progression free survival could not be calculated because patients who experienced disease progression did not amount to 50% of all patients. Univariate analysis of clinical factors showed that patients with bile duct or pancreatic duct invasion (p=0.031), tumor size larger than 20 mm (p=0.036), liver metastasis (p=0.020), distant metastasis (p=0.005), lymph node metastacytopathosis (p=0.009), without resection of primary tumor (p=0.020), AJCC/UICC stage III/IV (p=0.020), or ENETS stage III/IV stage (p=0.009) were more likely to demonstrate shorter PFS (Table 3). Multivariate analysis for prognostic factors demonstrated that bile duct or pancreatic duct invasion [p=0.010, hazard ratio (HR)=95.046], and tumor location (non-head portion of pancreas) (p=0.036, HR=7.381) were significant factors for predicting shorter PFS (Table 4) (Fig. 3).
Treatment modality
Surgical resection was performed in 27 patients. Among them, R0 resection was performed in 24 patients, all of which were still alive without recurrence. Surgical methods consisted of pylorus preserving pancreatoduodenectomy (n=7), distal pancreatectomy (n=9), enucleation (n=8), central pancreatectomy (n=1), total pancreatectomy (n=1), or wedge resection (n=1). The other treatment options that were performed included concurrent neoadjuvant chemoradiotherapy (n=3), palliative chemotherapy (n=6), transarterial chemoembolization (n=2), transarterial chemoinfusion (n=1), or somatostatin analog (n=3).
DISCUSSION
Although 64.3% of gastroentero-pancreatic endocrine tumors present metastasis at diagnosis, the 5-year survival is up to 77.5%.10 For pancreatic sites, poor differentiation and distant extra-hepatic metastasis have been reported as major negative prognostic factors.10 Han, et al.7 reported that large PNETs, regardless of their functional status, were more likely to be associated with malignancy and a predictor of worse survival. Paik, et al.8 demonstrated that resection of primary tumors in patients with PNET was associated with improved survival regardless of tumor stage. Kang, et al.9 reported that non-functioning tumors were more likely to show recurrence. In our report, patients with liver metastasis or without resection of primary tumors had shorter overall survival. Patients with bile duct or pancreatic duct invasion or tumors located in the body or tail of the pancreas were more likely to demonstrate shorter PFS. We assumed that tumors located at the body or tail of the pancreas are less likely to present symptoms, so that tumors are found at advanced stages and are more likely to show disease progression. But, in our data, there was no definite difference in tumor stages between the two groups [ENETS stage III, IV: 40% (head) vs. 41.7% (non-head)].
As for diagnostic tools of PNETs, Chromogranin A appears to be the most useful serum marker for the diagnosis, staging and monitoring thereof.11 Chromogranin A is gaining acceptance as a serum marker for neuroendocrine tumors.12 In our report, almost all patients were diagnosed by positive chromogranin A staining, but serum tests for chromogranin A was not performed. EUS is of high value for localizing primary lesions, and EUS-guided FNA can accurately diagnose and predict prognoses based on cytopathologic examination with immunocytochemistry. Somatostatin receptor scintigraphy is a very sensitive procedure for diagnosing gastrinomas but not insulinomas. Computed tomography, ultrasonography and magnetic resonance imaging are primarily useful for visualizing metastasis, which are able to predict prognoses.13 Cholangiography can be useful for evaluating bile duct invasion. Cholangiography was performed and was helpful in recognizing bile or pancreatic duct invasion, which was confirmed as a significant prognostic factor in our study. Although EUS-guided FNA was performed, it was not helpful in predicting prognoses, because most of patients' data did not include Ki-67 index and mitotic rate. Further evaluation, such as mitoses measurement and Ki-67 labeling, will be needed for predicting more reliable prognoses.
Previous classification systems make a distinction between low and high-grade malignant NETs, but could not differentiate further prognosis. In contrast, according to tumor stage and Ki-67 index, new TNM classification can differentiate prognosis significantly.14 Also, according to size, mitoses, invasiveness, and Ki-67 labeling, recent WHO classification classifies PNETs into grades 1 (neuroendocrine neoplasm, low grade), 2 (neuroendocrine neoplasm, intermediate grade), and 3 (neuroendocrine carcinoma) in an attempt to predict natural history from pathology reports.4 In 2006, Rindi, et al.15 introduced a four stage TNM classification for PNETs, which has subsequently been adopted by the ENETS. In 2010, the new AJCC/UICC TNM staging for PNETs was proposed distinguishing between localized tumors (stage I), locally advanced resectable tumors (stage II), locally advanced unresectable tumors (stage III), and distantly metastasized tumors (stage IV).16 In our study, we were able classify PNETs by AJCC/UICC TNM staging, but we could not classify PNETs based on histologic factors (WHO classification) due to insufficient data. From now on, PNETs need to be diagnosed based on both AJCC/UICC TMN staging and histologic findings (tumor differentiation, Ki-67 index, mitotic rate) through detailed data collection.
In contrast to our study, some articles reported that a small and pathologically benign nature did not predict a good prognosis in PNETs, so curative resection should be considered initially, even in cases of incidental PNETs. Even in patients with PNET metastatic lesions, resection of primary tumors should be considered for reasonable operative candidates.17 In particular, resection of primary PNETs tushould be given to patients with treatable hepatic metastasis. Aggressive surgical resection for select individuals with PNETs can be performed safely and may improve both symptomatic disease and overall survival. Prognostic indices such as tumor differentiation and the ability to achieve R0/R1 resection have been prognostic factors in PNETs and should be considered when planning aggressive surgical management.18 Surgical strategy for PNET depends on the size and location of the tumor as well as the risk of malignancy. Selection of the proper surgical method is important to preventing postoperative complications and recurrence. Radical resection including primary and metastatic lesion may improve survival of malignant PNETs.19 Only patients without distant metastasis underwent surgical resection in our study. Through analyzing prognostic factors, aggressive surgical resection can be considered even in cases of distant metastasis.
Transarterial chemoembolization, peptide receptor radionuclide therapy (PRRT), systemic chemotherapy, and radiotherapy can also be treatment options. PRRT uses somatostatin analogs to convey radioactivity within the tumor itself through somatostatin receptors. New biological agents and somatostatin tagged radionuclides are also under investigation20 as antiproliferative agents capable of stabilizing tumor growth in patients with metastatic PNETs.21 Advances in therapy and cooperative multicenter studies are needed to improve diagnosis, treatment and survival of patients with PNETs.22 Various treatment options besides surgery were tried in our study. Concurrent chemoradiotherapy was performed preoperatively in patients with lymph node metastasis or vascular invasion. Palliative chemotherapy (Etoposide, cisplatin) was performed in patients with distant metastasis. Transarterial chemoembolization was performed in patients with liver metastasis where surgical resection was not feasible. Somatostatin analogs were tried in patients with carcinoid syndromes such as flushing, diarrhea, and abdominal pain.
Prognostic factors were inconclusively reported in previous reports. We analyzed prognostic factors for predicting survival and disease progression in order to predict preoperative prognostic factors and postoperative results. From our report, resection of primary tumors can improve survival, and pancreatic or bile duct invasion, tumor location (body or tail of pancreas) were poor prognostic factors for disease progression. However, our study has some limitations, including a very wide confidence interval, due to small number of patients, and insufficient data for WHO classification.
In conclusion, patients with bile duct or pancreatic duct invasion or tumors located at non-head portions of the pancreas or without resection of primary tumor should be monitored carefully. But, because PNETs is a rare subgroup of tumor, we need more time and enough histopathologic data such as mitotic count and Ki-67 index for reliable prognostic prediction.




 XML Download
XML Download