

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hepatitis B virus (HBV) is a small relaxed-circular partially double-stranded DNA (3.2 Kb) virus belonging to the Hepadnaviridae family. Hepatitis B is a global health issue and represents an enormous health burden. Despite the availability of an effective vaccine, more than 350 million people are infected with HBV worldwide. Chronic hepatitis B (CHB) is the 10th leading cause of mortality worldwide, with more than 1 million deaths annually attributed to CHB-associated complications, such as liver cirrhosis and hepatocellular carcinoma (HCC).1,2 The natural course of HBV infection and the development and progression of CHB is determined by a plethora of contributing factors, which typically combine to influence disease severity, responsiveness to antiviral therapy and clinical outcome. There is a complex interplay between host and virus factors which influence the natural history of CHB and disease progression, including: age at infection; gender; route of infection; HBV genotype and particular virus mutations. These are extensively reviewed in Kim, et al.3 HBV is considered a non-cytopathic viral infection of hepatocytes. The liver damage associated with CHB is attributed to the host immune response to the infection. The clinical course and liver disease outcomes following HBV infection varies on an individual basis, possibly reflecting the intricate virus-host interplay. HBV infection in adulthood presents as an acute infection which is rapidly cleared in 90-95% of cases. Conversely, over 90% of perinatal or early childhood HBV infections result in the development of CHB.4-6 In CHB, an asymptomatic period of 20-30 years is followed by the development of liver cirrhosis leading to HCC and death in over 25% of patients. Viral persistence and the development of CHB has been associated with viral manipulation and evasion of the host's immune system, and the establishment of host "immune tolerance", which has lead to HBV being qualified as a stealth virus. A key viral tolerogen is the precore protein or hepatitis B e antigen (HBeAg), which is reported to attenuate the host immune response to the nucleocapsid protein,7 down-regulate and manipulate the innate and adaptive immune responses,8 and traverses the placenta to induce immune tolerance in utero thereby promoting persistence following perinatal infection.9 HBV associated HCC rates are rising rapidly which in themselves constitute an enormous health care burden. Current treatments for HBV infection are susceptible to acquired drug resistance mutations (antiviral agents such as neucleos(t)ide analogues) or exhibit poor responder (approximately 30% patients) rates (immune modulators such as Interferon). The development of new therapeutic approaches, potentially targeting and regulating the HBV precore protein to alleviate immune tolerance, is necessary to improve clinical outcome following HBV infection and circumvent the development of CHB.
MOLECULAR PATHOGENESIS OF HBV
The HBV genome encodes five viral proteins translated from mRNA transcripts, which are encoded by four overlapping open reading frames (ORFs). These are: the envelope (there are three surface protein sizes) or hepatitis B surface antigen (HBsAg); polymerase (pol), hepatitis B x antigen (HBxAg); nucleocapsid or hepatitis B core antigen (HBcAg); and the precore or HBeAg. The virus replicates in the cytoplasm of hepatocytes via the endogenous viral-encoded polymerase performing reverse transcription of the packaged pregenomic RNA template contained within the viral nucleocapsid. Following first strand (negative DNA) synthesis, core particles are enveloped and virions secreted through the cellular Endoplasmic Reticulum (ER) & Golgi compartments. HBV can persistently infect the liver and HBV chronicity or CHB is defined as persistent HBV infection (HBsAg positive) for greater than 6 months.10 Several studies have suggested that the PreCore-Core (preC-C) gene, which encodes both HBeAg and HBcAg, plays an important role in establishing persistent HBV infection.11 Interestingly, its location within the HBV genome ensures that it is the first gene transcribed and translated.
HBV can be classified into 10 genotypes (A-J), based on a genome sequence divergence of greater than 8%, and further classified into sub-genotypes which diverge by 4-8%.3,12,13 HBV genotype plays a role in determining disease progression and clinical outcome, indicating sequence specific biomarkers for virulence within the HBV genome. HBV genotypes B and C correlate generally with a poorer prognosis, conferring a greater likelihood to progress to CHB and the development of HCC (Table 1). Furthermore, genotypes B and C display a poorer response to interferon (IFN) treatment, in comparison to genotype A, whilst genotype D, a predominantly European genotype, has the poorest response.13 HBV genotypes B and C are predominantly geographically localised to Asia (particularly China), where HBV is endemic in the population, and there is a high prevalence (10-20%) of CHB.13 Subject age at the time of HBV infection inversely correlates with the risk of chronicity. In China, where HBV genotypes B and C are prevalent, the most common route of infection is via perinatal transmission, not withstanding a schedule of vaccination and hepatitis B immunoglobulin (HBIg) prophylaxis from birth. The likelihood of developing CHB following the perinatal HBV transmission infection route is enhanced by the presence of HBeAg in the mother. There is a >90% transmission rate to babies born to mothers who have HBeAg positive CHB (in the absence of vaccination and prophylaxis), and 90% of these perinatal HBeAg positive infections develop CHB.4-6 Indeed, HBeAg is required for viral persistence and the development of chronic disease, and genotypes B and C HBV, which are endemic to China, are associated with longer HBeAg positive status before seroconversion to anti-HBe, up to 2-3 decades longer than the European genotypes A and D HBV, where the risk of HCC progression and mortality due to HBV is far lower (Table 1). HBeAg positivity is a strong predictor for HCC.14 Furthermore, HBV genotypes B and C, or HBeAg positive infection, has been associated with more efficient inhibition of the innate immune response thereby promoting viral persistence.15
The natural course of HBV infection can be considered as five separate phases: immune tolerance; immune clearance; inactive/non-replicative; reactivation; and immune control.3,16 However, many factors, host and viral, affect the course of disease and the clinical prognosis.17 A longer immune tolerant phase is associated with genotypes B and C, or more specifically to cases of perinatal transmission, which is common for genotypes B and C, where it can persist for several decades. This phase is defined by HBeAg and HBsAg positivity, relatively normal alanine aminotransferase (ALT) levels, and minimal liver damage in spite of very high levels of HBV DNA within the blood and liver. However, during immune tolerance the viral DNA integrates into the host genome, increasing the risk for the development of HCC in later years. Indeed, HCC development is also linked to the length of infection, another reason for the high incidence of HCC in genotype B and C HBV. In the immune clearance phase, also known as HBeAg positive CHB, activation of host immune responses occurs, and is associated with increased ALT levels, liver inflammation and hepatocyte death. Although it is unclear how the immune response is triggered, this phase eventually witnesses the seroconversion of HBeAg positive to anti-HBe positive status (HBeAg seroconversion), that is, the presumed loss of the HBeAg immunotolerant effect, which is potentially induced by immune pressure favouring the emergence of dominant precore or basel core promoter (BCP) mutants. The markers of the inactive phase are the return to normal ALT levels, and the continued HBeAg negative/anti-HBe positive status, and very low or undetectable HBV DNA within blood. Patients in the inactive phase can revert back to HBeAg positive CHB (immune clearance). It is important to note that the diagnostic determination of HBeAg status in fact reports on HBeAg 'in excess' in the serum, and actually HBeAg or anti-HBe negative status may not be absolute, but rather in an immune complex equilibrium. The reactivation phase, also called immune escape or HBeAg negative CHB, is associated with increased ALT levels and liver inflammation, and the appearance of precore/core BCP and/or G1896A precore stop codon mutant viruses, the later of which is more dominant in genotype D virus, and therefore the European and African regions. Some patients (1% per annum) progress to immune control, seroconverting from HBsAg to anti-HBs, and exhibiting normal ALT levels. However, HBV DNA may remain present in the liver as cccDNA, indicative of occult HBV infection, which exerts oncogenic effects associated with HCC development.17
IMMUNE RESPONSE TO HBV
The clinical outcome of HBV infection is influenced by numerous virus and host specific factors, including; HBV genotype, infection route, age at infection, and gender.3 HBV infection of hepatocytes is considered non-cytopathic, with liver damage associated with CHB the result of continual attempts by the host immune system to clear the virus from the liver, hence, the disease process. HBV infection in adults usually presents as an acute, self-limiting infection, with viral clearance achieved in 90-95% of cases. HBV clearance is dependant on the strength and specificity of the adaptive immune response, specifically the T cell response, although B cells are involved in presentation to CD4+ T cells and produce neutralising antibodies.18 Healthy adults with a mature immune system are better placed to achieve viral clearance, in comparison to children and babies (perinatal or horizontal transmission routes), whose immune system is still developing and therefore more receptive to HBV induced tolerance and manipulation of the immune response. Indeed, over 90% of perinatal or early childhood (<5 years) HBV infections result in the development of CHB,4-6 which is associated with the development of liver cirrhosis leading to HCC and death in over 25% of patients. The progression of CHB to clinical outcomes is preceded by asymptomatic disease, lasting 2-3 decades, during which time HBV effectively evades the immune response and clearance, through tolerance of the innate immune system and as a consequence, a weakened adaptive response.
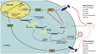
The innate immune response constitutes the first line of host defence, responsible for priming and activating the adaptive immune response. The innate system targets exogenous pathogen infection through the recognition of pathogen-associated molecular patterns. The Toll-like receptors (TLRs) are the primary pathogen pattern recognition receptors involved in the innate response, and signal via Toll/IL-1 receptor (TIR)-mediated pathways to promote pro-inflammatory cytokine responses. The TIR-signalling domain of TLRs is highly conserved and initiates the intracellular signalling cascade.19 The TLR (TIR) signalling process ultimately results in pro-inflammatory cytokine production and activation of NF-κB pro-inflammatory transcription mediators, which affect the later adaptive response.20,21 Strategic blocking of the innate immune system, including the receptor and cascade signalling/adaptor molecules, is a common pathogenic mechanism for host immunity evasion, and pathogen survival and persistence.22 Indeed, the HBeAg of HBV immunomodulates the innate immune response through interaction with TLR2 and several TIR domains (Mal, TRAM) to suppress signalling cascades and down-regulate downstream inflammatory factors such as NF-κB.15,23-26 In vivo HBeAg positive status can be associated with low pro-inflammatory cytokine state (i.e. low TNF levels), suggestive of a state of suppressed innate immunity (Fig. 1). Despite the significant overlap of HBeAg and HBcAg, the HBeAg encodes a unique N-terminal 10 mer, which is highly evolutionarily conserved across all orthohepadnaviruses.24,27 The HBeAg 10-mer has recently been shown to contain a TIR domain mimic,24 potentially enabling manipulation of innate immunity TLR/TIR signalling cascades, towards promoting host immune tolerance.
The host adaptive immune response to HBV incorporates T and B lymphocytes, which are activated by antigen presenting cells following presentation of specific viral protein components. The HBeAg inhibits T cell activation via down-regulation of the expression of the CD86 co-stimulatory molecule on Kupffer cells and CD14-positive monocytes,15 thereby blocking stimulation and subsequent maturation of Th0 cells. The mechanism of action of activated CD8+ T lymphocytes is to kill HBV infected hepatocytes, which has the unfortunate side-effect of causing liver damage. The B lymphocytes produce the antibody response against key viral antigens, including the production of neutralising anti-HBs antibodies to clear serum virions and prevent re-infection, and an anti-HBc and anti-HBe antibody response to mop up viral proteins. The IgG secreting B lymphocytes or antibody secreting plasma cells response is the target of the HBsAg based HBV vaccine, with the antibody response peaking at day 7 post-vaccination, and constituting 6% of blood B lymphocytes. The memory B lymphocyte response, conferring long-term anti-HBs HBV immunity, peaks by day 14-21 post-vaccination, and constitutes 1% of blood B lymphocytes.28 Anti-HBs antibodies also constitute the basis of the prophylactic HBIg treatment. The HBIg formulation constitutes anti-HBs isolated from high titre sera and is administered intravenously or intramuscularly to prevent HBV infection following exposure, including those individuals such as liver transplanted HBV positive patients, perinatal exposed infants of HBV positive mothers, acute exposure to HBV positive blood, sexual exposure, and household exposure to acute HBV infected persons.29 HBIg has been shown to be internalised via the FcRn receptor on the hepatocyte surface, and subsequently to enhance intracellular retention and degradation of HBsAg, thereby reducing circulating HBsAg and HBV virions.30,31 However, HBIg has been shown to exert HBsAg selection pressure and promote the development of HBsAg immune escape mutant virus, most commonly the G145R HBsAg mutant. The B cell antibody response also targets HBcAg and HBeAg, with seroconversion to anti-HBe considered a marker of transition into the inactive phase of disease. Patients can see-saw between HBeAg positive and anti-HBeAg positive, which is likely due to a fluid state of HBeAg and anti-HBe production, some breakthrough infection, the formation of immune complexes, and/or detection of the HBeAg or anti-HBe in excess at a given sample/timepoint.32 Which is to say, patients may be HBeAg positive far longer than reported, or conversely anti-HBe positive earlier than reported, as immune-complexed HBeAg or anti-HBe are not 'available' to be detected in current diagnostic binding assays, which can detect only the antigen component in excess.
PRECORE PROTEIN/HBeAg: THE IMMUNOMODULATOR
HBV persistence and the establishment of CHB is regulated by the precore protein or HBeAg.33 The precore protein (HBeAg) is a secreted (and hence soluble) accessory protein form of the core protein, which is not required for replication but which appears to attenuate host immune response to the intracellular nucleocapsid protein.7,8 The HBeAg is therefore considered an important tolerogen since it contributes to HBV persistence in the infected host, possibly functioning as an immune tolerogen in utero as the secreted, soluble HBeAg can cross the placenta.9 Furthermore, in animal models HBeAg has been shown to regulate the host's immune response, but the cellular processing of the precore protein is complex.34
The HBeAg and HBcAg genes are transcribed from separate although highly homologous RNA transcripts. Accordingly, the two proteins share significant (more than 90%) amino acid identity.35 The mature intracellular HBcAg or nucleocapsid protein (p21) of approximately 21 kDa (183 residues) includes a C-terminal arginine-rich DNA binding domain for packaging the viral genome. PreC-C gene translation for HBeAg produces the precore precursor (p25) protein (Fig. 1). An N-terminus, 29 residue ER signal sequence directs the precore precursor to the ER for two-step processing. From the N-terminus, 19 residues are cleaved,36 producing p22 intracellular precore protein. Following this, approximately 34 residues encoding the nucleocapsid-associated arginine-rich protamine domain are cleaved from the C-terminus by a furin-like protease.37-39 The exact site of concleavage remains contentious and is also influenced by genotype specific sequence differences in this region of the precore/core sequence.27,40,41 This second processing event produces the HBeAg protein (p17), which is a secreted protein of 159 residues or approximately 18 kDa.34 The precore protein shares 149 common residues with the core protein, but importantly possesses a unique N-terminal hydrophobic and cysteine-containing 10 mer, which confers precore specific structural, serological and immunological characteristics in comparison to the p21 core protein. The HBeAg is secreted and evident in serum as a soluble dimeric protein (Fig. 1). Intracellular precore forms DNA negative capsids due to the lack of the C-terminal arginine rich DNA-binding domain, which can be enveloped and released as DNA-negative 'decoy' Dane particles.37 Precore capsids are less stable and regular than core nucleocapsid, due to the unique 10 mer, which disrupts the core specific C61-C61 stabilising dimer/capsid bond, in preference for a C-7-C61 bond in HBeAg dimers/capsids.41 However, the HBeAg retains capsid formation ability even following substitution of all cysteine residues for alanine, albeit these cysteine deficient precore capsids are less stable.42 Additionally, the precore unique 10 mer encodes a TIR like domain, to potentially enable the precore protein to mimic and manipulate the innate signalling pathways,24 and tolerise the host immune response to HBV, thereby enabling the viral persistence and the development of CHB.
Little is known about the function of HBeAg in the viral life-cycle.43 It has been observed that in transfected cells an increase in the expression of the preC-C gene leads to inhibition of HBV replication,44 whilst mutations (e.g. G1896A) leading to the abolition45 or reduction of preC-C gene expression result in a significant increase in HBV replication.46,47 This inverse correlation between precore protein expression and HBV DNA replication fits comfortably with the modulatory role of precore protein, whereby dampening viral replication promotes host immune response evasion and enables viral persistence and the establishment of CHB.48
The precore protein manipulates and dampens both the innate and adaptive immune responses to enable the establishment of CHB. HBeAg is the soluble, secreted form of the viral nucleocapsid (or core) protein, and these proteins share over 90% protein sequence homology, but not immunological B-cell (antibody) epitopes.32 The precore protein is reported to down-regulate cellular genes responsible for intracellular signaling, essentially functioning like a tumour suppressor protein with anti-apoptotic activity.48 Further more, precore protein dampens the host immune response to viral infection via down-regulation of innate immune receptors, specifically TLR2.15,25 Indeed, HBV replication is associated with the upregulation TLR2 signaling pathway in the absence of the tolerogenic HBeAg.15 Thus it is possible to speculate that HBeAg positive CHB is associated with a low TNFα or low pro-inflammatory innate-suppressed state, whereas HBeAg negative CHB is associated with a high pro-inflammatory or high TNFα state (Fig. 1). This results in contrasting phases of CHB pathogenesis between HBeAg positive and negative disease. HBeAg manipulation and downregulation of the TIR-dependant innate immune response may allow HBV to persist and escape immune clearance. Interestingly, the unique HBeAg N-terminal 10 mer is highly conserved across the HBV genotypes, and is found in all mammalian-infecting Hepadnaviridae.27 This highly conserved precore sequence incorporates a putitative box-2-like TIR motif (KLCL) and enables HBeAg to antagonise the TLR signalling cascade, thereby allowing the virus to evade the innate immune response.24 HBV genotype specific sequence variation within HBeAg, but outside the conserved N-terminal 10 mer, confers more efficient inhibition of the TIR-dependant innate immune response in genotype B and C virus, as opposed to genotypes A and D (unpublished data). Yet more evidence indicating the importance of the precore effect on the immune response, considering that HBV genotypes B and C are associated both with poorer clinical outcome and longer period of HBeAg positive infection.
TREATMENT OF CHB
Currently there are two therapeutic approaches available for the treatment of CHB; these are antiviral agents (nucleos(t)ide analogues) and immune-based therapies (IFNα or Pegylated-IFNα).49 Antiviral agents, or nucleos(t)ide analogues (NA's), target the HBV polymerase and therefore viral DNA replication and include the drugs: Lamivudine (LMV); Adefovir (ADV); Entecavir; Telbivudine; and Tenofovir (TFV). These drugs are associated with high costs, very long durations of therapy, and are susceptible to the development of acquired drug resistance. They require careful patient management and compliance to maintain effectiveness and limit the development and selection of drug-resistant mutant virus. This is particularly important for HBV, which is a small and efficiently packaged virus genome consisting of multiple overlapping ORFs. This results in the HBsAg completely overlapping the polymerase gene, and means any acquired drug resistant mutations in Pol typically have an equivalent associated effect on HBsAg, potentially introducing HBsAg mutants which could increase viral fitness, thereby promoting immune or vaccine escape. Indeed, the rtA181T mutation associated with LMV, ADV and TFV resistance results in a sW172 stop mutation in the overlapping HBsAg. The resultant truncated HBsAg has been claimed to be associated with increased risk to HCC progression,50 and is more frequently selected in genotypes B and C.51
Immune-based therapies such as, IFNα or PEG-IFNα, act to stimulate the immune response via modulation of T and B lymphocytes as well as some modest but direct antiviral activity to reduce viral protein transcription. However, IFNα therapy is associated with high costs, significant side effects, and poor treatment response, effective in only 30-40% of CHB patients. In the liver transplant setting HBIg is used to prevent graft re-infection.29 This approach has also been broadened to include treating CHB in restricted immune phenotype patients.52,53 In general terms, the precore protein exerts a negative effect on HBV immune-based treatment (e.g. IFNα) effectiveness, with higher HBeAg titres associated with poor treatment response rates. Patients who are responsive to current treatments, such as IFNα, have significantly lower pre-treatment baseline levels of circulating precore or HBeAg in serum than non-responder patients.54 There is no cure for CHB. An enormous unmet need exists (estimated at approximately 36 million patients) for alternative therapeutic strategies targeting early stage CHB prior to liver damage.55 The precore protein or HBeAg is a key viral protein that regulates host immune tolerance, viral persistence, and is essential for the establishment of CHB.33 The precore protein is an attractive target for the development of a new class of therapeutic biologicals with activity against HBeAg. It is hypothesised that treatments to regulate and control precore protein levels can improve clinical outcome. Reduction of the HBeAg burden and therefore dampening the HBeAg tolerogenic effect would enable up-regulation of the immune response and additionally, should increase the effectiveness of current antiviral treatments (e.g. IFNα). This concept provides a rational basis for the development of anti-HBe therapeutics in patient management to reduce serum HBeAg load, with the goal of interruption of the natural history of CHB, including blocking the progression of CHB to the main clinical end-points of liver cirrhosis and HCC.
ANTI-HBe THERAPEUTICS
Immunoglobulins are centrally important for the modulation of the immune response, and immunoglobulin therapy is already an accepted and clinically proven therapy in HBV treatment, with HBsAg targeted by HBIg therapy in the prophylactic vaccine and post-liver transplantation setting. To address the therapeutic target of reduced serum HBeAg load, the development of novel HBeAg immunoglobulin-based therapeutics could be explored to mimic and supplement the host anti-HBe response, leading to serum HBeAg being 'mopped up', or preferentially, for HBeAg to be retained intracellularly and degraded. An anti-HBe immunoglobulin treatment could effectively reduce HBeAg loads to relieve the tolerogenic effect on the innate immune response, and drive a positive upregulation of the host's endogenous response to HBV infection. These outcomes would enhance the success rate of current IFNα treatment, which is more effective when HBeAg pre-treatment titres are low. Our research has seen the isolation and development of a novel shark new antigen receptor antibody (IgNAR) variable domain (VNAR), which targets the HBV precore protein. IgNARs are bivalent, targeting antigen through a single VNAR (approximately 14 kDa) displaying two complementarity-determining region (CDR) loops.56,57 The VNAR antibody format possesses advantageous capabilities of compact size and robust stability. VNAR's also display cryptic antigenic epitope access via unusually long and variable CDR-3 loops.58-62 We isolated an HBeAg-specific VNAR (known as H6), which we subsequently characterised as specifically targeting a unique and conformationally dependant HBeAg epitope, at mid-nanomolar range binding affinity, through a long (18 residue) CDR3 loop.35 No existing monoclonal antibody has previously exhibited this particular property. Further development and characterisation has been undertaken, and the isolated anti-HBe VNAR domain validated as an ER-localised intracellular antibody (intrabody). The anti-HBe intrabody was found to: 1) specifically and effectively bind precore protein (and not core protein) intracellularly; 2) disrupt precore processing through inhibition of precore aggregation, or promotion of ER degradation/processing of precore to circumvent aggregation; 3) and reduce HBeAg secretion down to 0.3 fold in Huh7 in vitro cells models.35 These findings indicate that the anti-HBe intrabody may nupossess therapeutic potential. Additional development of the anti-HBe VNAR domain would entail engineering with a human Fc domain, for intracellular uptake via the hepatocyte FcRn immunoglobulin receptor. As detailed earlier, immunoglobulin regulation of HBeAg has therapeutic potential, targeting intracellular HBeAg to reduce serum HBeAg load, subsequently alleviating or breaking immune tolerance, and increasing the effectiveness of current antiviral treatments, such as IFNα.54 Indeed, anti-HBe polyclonal antibody treatment has also been shown in vitro to prevent HBeAg-induced down-regulation of the innate (TNFα) immune response.15 HBV immunoglobulin treatment is already standard of care in the form of HBIg, which entails administration of HBV surface antigen (HBsAg) specific antibodies, to effect a reduction in viral load and serum HBsAg following liver transplantation, and blocking perinatal exposure in infants of HBV positive mothers. Furthermore, research has demonstrated that HBIg antibody is internalised into hepatocytes via the FcRn cell surface receptor, and subsequently co-localises with HBsAg, to reduce envelope formation and virion secretion,31 which supports the concept of targeted intracellular antibody delivery to hepatocytes for intracellular retention of viral proteins. We propose that an immunoglobulin anti-HBe therapeutic based on the development of novel immunoglobulin domains, functioning intracellularly, to target HBeAg reduction, will provide an effective therapeutic option for the treatment of CHB. The therapeutic goal is to avoid or slow the development of CHB-associated complications.
CHALLENGES OF HBV MODELS
Good models, both in vitro cell culture and in vivo animal models, for HBV and particularly CHB, are a significant barrier for research and pre-clinical therapeutics testing. Models of HBV are found lacking, and not generally biologically relevant for a variety of reasons, which include: non-permissive to HBV infection or replication; no development of HCC; milder disease outcomes; lower progression rates to CHB; dissimilar routes of transmission; and virus sequence variation.63
In vitro cell culture models of HBV include hepatocyte cell lines such as Huh7, HepG2.2.15 and AD38, which can be transiently or are stably transfected to express HBV proteins or infectious virus.48,64,65 However, these immortalised cells lines, whilst useful for research purposes, have lost the ability to be permissive to HBV infection and viral replication, lack integral cell signalling and immunity pathways, and do not adequately reflect disease state in terms of analysing therapeutic treatments. Primary human hepatocyte cultures, whilst more relevant for studying HBV replication and signalling cascade events, are inherently difficult to maintain and quickly become non-permissive to HBV infection. There are many animal models in use for the study of HBV, although none truly reflect HBV infection and clinical disease development in humans. Although mice are not susceptible to HBV, are not permissive to HBV infection and replication, and possess fundamental immunological differences to humans,66 several mouse models have been engineered for the study of HBV. The humanised SCID mouse model or HBV-Trimera mouse are a CB6F1 stain which undergo total body irradiation, followed by reconstitution with either human lymphocytes to produce human antibodies, such as humanised HBV anti-HBs specific monoclonal antibodies (XTL-17 and XTL-19);67 or conversely reconstituted with harvested SCID/NOD bone marrow cells and transplanted with ex vivo HBV-infected human liver fragments, to investigate HBV viremia and test HBV therapeutics.68,69 These HBV-Trimera mice are viremic for approximately 1 month, peaking at day 18-25, which allows for an approximately 20 day window for therapeutics testing. It is worth noting however, that the transplanted human liver represents hepatocyte survival but not growth, so this is not strictly speaking a replicative model. Another engineered murine model are the Urokinase-type plasminogen activator mice,70 into which isolated human hepatocytes and spleenic cells are injected into 6-14 day old mice, followed by infection with human HBV serum 5 weeks post transplantation.
There are a number of popular larger animal models for HBV, including Tupaia, Ducks and Woodchucks.63 Primary Tupaia hepatocytes are harvested for the study of transient HBV infection.71 Ducks have long been used in HBV research; however, whilst duck HBV (DHBV) is similar to human HBV, there is no associated liver disease or HCC. Woodchuck HBV (WHV)72 displays significant genomic variation compared to human HBV, with greater than 30% sequence variation between WHV HBeAg and human HBV HBeAg, possibly eliminating key immune recognition epitopes.27 Furthermore, the Woodchuck immune system is poorly characterised, which is problematic for extrapolation of data to humans. Primate models of HBV certainly reflect human HBV more closely; however there remain numerous, now mainly ethical barriers for use of primate models. The chimpanzee model of HBV displays similar infectivity to human HBV, however, the disease is milder and vertical route of HBV transmission, which is common in humans and associated with CHB development, is rare in chimps, and this is reflected by the low (<5%) rates of CHB in chimps.73 Furthermore, Chimpanzee's infected with HBV are both enormously expensive and now not available (especially chimps with chronic infection), due to strict ethical constraints. There are fundamental differences in immune responses between humans and chimps, and accordingly chimps exhibit milder disease and minimal hepatitis in chronic infection, with no recorded cases of HCC.66 The chimp model is therefore of questionable usefulness in the development of therapeutics to treat CHB. Indeed, there is no perfect model for human HBV available for the testing HBV therapeutics and studying HBV replication and immune signalling cascades. As a result much data must be extrapolated from in vitro and in vivo models to best fit the human HBV, often resulting in confounding conclusions.
CONCLUSIONS
The HBV virus is a potent and effective manipulator of the host immune response, allowing the virus to survive and successfully persist in the host for decades. Progression to CHB results in 90% of perinatal and childhood infections, with 25% of these patients suffering the severe clinical outcomes of liver disease and HCC. Viral persistence and the establishment of CHB is driven by the precore protein or HBeAg, and its immunomodulatory effects on the innate immune system. The HBeAg tolerogen has been linked to down-regulation of innate immune receptors, such as TLR2, and antagonises innate signalling cascades to induce a suppressed inflammatory state. In addition to this, HBeAg levels affect, and are a predictor of, IFNα treatment response, with higher baseline HBeAg levels associated with poor IFNα treatment response. Interestingly, HBeAg levels and extended periods of HBeAg-positive infection are associated with the HBV genotypes B and C, which are in turn associated with more efficiently inhibited innate immunity responses, poorer responses to IFNα treatment, and poorer disease prognosis. Taken together, these facts highlights the HBeAg as a potential target for therapeutic regulation towards improved clinical outcome, response to IFNα treatment, and disruption of the progression to CHB. The therapeutic reduction of a viral protein to treat HBV is clinically accepted for HBsAg, using HBIg, an anti-HBs immunoglobulin treatment. A similar approach could be achieved for HBeAg reduction. Here we highlight an anti-HBe intrabody (H6), which has been shown to possess highly selective intracellular activity targeting HBeAg, reducing HBeAg secretion and promoting intracellular degradation. Potentially, this anti-HBe intrabody could be developed as a humanised immunoglobulin treatment to therapeutically reduce serum HBeAg titres, and achieve the goals of alleviating immune tolerance, and improving the success of IFNα therapy. Novel HBV targets and the development of new therapeutics are necessary for the treatment of CHB, to reduce the progression to liver disease and HCC, and reduce the global health burden of HBV infection.
 XML Download
XML Download