

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Oral cancers are aggressive tumors arising in the lip, tongue, floor of the mouth, gingivae, palate, buccal mucosa/vestibule or salivary glands, and are associated with a high mortality rate.1,2 Over 300000 cases of oral and pharyngeal cancer are annually reported worldwide with male outnumbering female patients.3 Oral cancer in Korea, based on the 2008 report of the national cancer center, has an incidence of 2.6 cases per 100000. The treatment of oral cancer typically involves chemotherapy in combination with other modalities such as radiation therapy or surgery.4 However, the mechanisms underlying tumor generation in the oral cavity are not yet fully understood, and the mortality rate for this disease has not improved in recent years.
Capsaicin, N-vanillyl-8-methyl-α-nonenamide, is a spicy component of hot pepper and comprises an aromatic ring and dipolar amide bond in its chemical structure. Recent reports have indicated that capsaicin has beneficial effects on certain types of cancers such as tumors of the colon, prostate, lung and breast.5-7 Capsaicin has also been shown to inhibit N-methyl-N'-nitro-N-nitrosoguanidine-induced gastric carcinogenesis and reduce the incidence of colonic adenocarcinomas in an azoxymethane-induced rat colon carcinogenesis model.8,9 In vitro studies of transformed cells and various types of cancer cells have further shown that capsaicin induces programmed cell death.10 These include human stomach cancer cells, hepatocarcinoma, glioblastoma and neuroblastoma cells.8,11-13 However, the molecular mechanisms underlying capsaicin-induced apoptosis are cell type dependent: capsaicin induces apoptosis in sensory neurons by increasing calcium influx and does so by activating vanilloid receptors in some transformed cells.14-16 In human colon cancer cells, capsaicin triggers apoptosis through the inhibition of plasma membrane NADH-oxidoreductase activity and/or NADH: coenzyme Q oxidoreductase in the mitochondrial electron transport system, generating reactive oxygen species.5,10,17 Moreover, capsaicin was found to be associated with PPARγ during the regulation of cell growth and apoptotic cell death in breast or colon cancer cells.18 Despite the cumulative evidence for the tumor suppressive effects of capsaicin, however, few studies have been undertaken to date on the effects of capsaicin on cell signaling and the molecular pathways leading to apoptosis in oral cancer cells.19 In the present study, we investigated the effects of capsaicin on FaDu human pharyngeal squamous carcinoma cells and demonstrated that capsaicin induced apoptosis in FaDu cells.
MATERIALS AND METHODS
Materials
Capsaicin was purchased from Sigma Chemical Co. (St. Louis, MO, USA). Minimum essential medium (MEM), fetal bovine serum (FBS), and antibiotics/antimycotics were purchased from Gibco BRL (Grand Island, NY, USA). Caspase activity was measured using a caspase cellular activity assay kit (Calbiochem, Darmstadt, Germany). 4, 6-Diamidino-2-phenylindole (DAPI) was purchased from Sigma. Polymerase chain reaction (PCR) primers were purchased from Bioneer (Daejeon, Korea). Antibodies were purchased from the following sources: cdk-1, cdk-4, cyclin B1, Bcl-2, Bad, Bax and all secondary antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA); cdk-2, cyclin D1 and PARPp85 from Epitomics (Burlingame, CA, USA). All other chemicals were obtained from Sigma.
Cell lines and cell culture
Human pharyngeal squamous carcinoma cells were purchased from the Korean cell line bank (KCLB, Seoul, Korea) and were maintained at 37℃ in humidified atmosphere at 5% CO2 in MEM supplemented with 10% FBS and antibiotics/antimycotics.
Growth inhibition
Growth inhibition was assessed via an MTT assay. Briefly, FaDu cells were plated at a density of 1×105 cells/well on 24-well plate. After overnight growth, the cells were treated with various concentrations of capsaicin for 24, 48 and 72 hours, with medium replacement every 24 hours. At the end of treatment, 30 µL of the tetrazolium compound MTT (Sigma, St. Louis, MO, USA), and 270 µL of fresh medium were added. After further incubation for 4 hours at 37℃, 200 µL of 0.1 N HCl in 10% SDS was added into each well to dissolve the tetrazolium crystals. Finally, the absorbance at a wavelength of 540 nm was recorded using an ELISA plate reader (Thermo Fisher Scientific, Waltham, MA, USA).
DNA fragmentation
Cells cultured in 100 mm dishes were treated with capsaicin (100 µM and 300 µM) for 24 hours, trypsinized and collected with ice-cold phosphate-buffered saline (PBS). After centrifugation at 300 g for 10 minutes at 4℃, the cells were washed with PBS and centrifuged again at 5000 rpm for 5 minutes at 4℃. Cell pellets were resuspended in 0.5 mL of lysis buffer (10 mM EDTA, 50 mM Tris-HCl, pH 8.0, 0.5% SDS, 0.5 mg/mL proteinase K) and incubated overnight at 50℃. The lysate was centrifuged at 14000 rpm for 5 minutes at 4℃ to separate the soluble DNA fragment from the intact chromatin pellet. The fragmented DNA was extracted with phenol/chloroform/isoamyl alcohol (25 : 24 : 1) and precipitated with ethanol. The purified DNA was treated with 200 µg/mL of DNase-free RNaseA for 1 hour at 37℃ prior to electrophoresis on a 1.8% agarose gel containing ethidium bromide.
Nuclear staining
Cells were incubated in 6-well plates with 200 µM capsaicin for 24 hours. After washing with PBS, the cells were fixed with 4% formaldehyde and dried briefly. The fixed cells were washed again with PBS, air dried and stained with the DNA-specific fluorochrome, DAPI. The cells were washed with PBS, air dried and mounted with 90% glycerol. The plates were then observed under a fluorescence microscope (Olympus Optical Co., Tokyo, Japan).
Flow cytometry analysis
Cells treated with various concentrations of capsaicin for 24 hours were harvested by brief trypsinization and centrifugation (at 300 g), washed in ice-cold PBS and fixed in 70% ethanol for 2 hours at -20℃. The fixed cells were then stained with PI (20 µg/mL propidium iodide, 200 µg/mL DNase-free RNase-A in PBS) for 15 minutes at 37℃. The DNA content of cells stained with propidium iodide was measured using a FACScan instrument and Cell Quest software (Becton Dickinson, Franklin Lakes, NJ, USA).
Analysis of cytochrome c release
The levels of cytochrome c in the cytosol were detected using an ELISA kit (Bender MedSystem, Wien, Austria). Cells were cultured in medium in the presence of 50, 100 and 200 µM of capsaicin for 12 hours. After treatment for 12 hours, the cells were lysed and centrifuged at 14000 rpm to obtain the cytosol fraction. The supernatants were transferred into a new tube and diluted 50-fold in an assay buffer in accordance with the manufacturer's instructions. The OD values were read on Multiskan EX Microplate Photometer (Thermo Fisher Scientific, Waltham, MA, USA), using 450 nm as a primary wavelength and 620 nm as a reference wavelength.
Cellular caspase-3 enzyme activity assay
Caspase activity in capsaicin-treated FaDu cells was analyzed using a caspase cellular activity assay kit, which includes human recombinant caspase-3, cell lysis buffer, an Ac-DEVD-pNA colorimetric substrate, a calibration standard (p-nitroaniline), a caspase-3 inhibitor and assay buffer. Caspase 3 activity in the cell lysates was determined colorimetrically in accordance with the manufacturer's instructions.
Reverse transcriptase-polymerase chain reaction (RT-PCR)
Cells were treated with 200 µM capsaicin for 24 hours. Total RNA was then isolated using gene plate from RNAture (Irvine, CA, USA). RT-PCR was performed to estimate the mRNA levels of anti-apoptotic Bcl-2 and pro-apoptotic Bad. The primer sequences used were 5'-AGGAGCTCTTCAG GGACGG-3' and 5'-CCAGGTGTGCAGGTGCC-3' for Bcl-2 and 5'-CCCGAGAGGTCTTTTTCC-3' and 5'-GCCTTGAGCACCAGTTTG-3' for Bax. For the detection of glyceraldehydes-3-phosphate dehydrogenase (GAPDH) transcripts, the primer sequences used were 5'-CCCAT CACCATCTTCCAGGAGC-3' and 5'-CCAGTGAGCT TCCCGTTCAGC-3'. The expected sizes of the PCR products were 161 bp (for Bcl-2), 109 bp (for Bax) and 473 bp (for GAPDH). The detected mRNA levels were normalized to the GAPDH using the Bio-Rad Image Master Program (Bio-Rad, Hercules, CA, USA).
Western blotting
Cells grown in 60 mm dishes (5×105 cells/dish) were treated with 200 µM capsaicin for 24 hours, harvested, washed with cold PBS (×2) and then lysed by the addition of 300 µL of lysis buffer [20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/mL leupeptin and 1 mM PMSF]. The cell lysates were homogenized by pipetting for 15 minutes on ice and then sonication for 1 minute. The supernatant from each lysate was collected into new tubes after centrifugation at 20000 g for 15 minutes at 4℃. Total protein concentrations were measured using the BCA™ protein assay kit (Pierce, Rockford, IL, USA). The protein bands were normalized to the beta actin expression level.
RESULTS
Effects of capsaicin on the morphology and viability of human pharyngeal squamous carcinoma cells
FaDu cells were treated in the presence or absence of capsaicin (200 µM) for 12 or 24 hours and the cell morphologies were analyzed under the microscope at different time periods. The density of the cells on the capsaicin-treated dish surfaces was reduced without change in shape when treated with 200 of capsaicin for 12 and 24 hours (Fig. 1A). To evaluate the effects of capsaicin on cell growth and proliferation, the cells (4×104/well) in 48-well plates were exposed to a series of capsaicin concentration for 24, 48, and 72 hours, and the percentage of living cells was determined via an MTT assay. Cells treated with 50, 100, 200, and 300 µM capsaicin showed an augmented decrease in cell growth as the capsaicin dose increased. In addition, the percentage of viable cells decreased as the incubation time increased. The observed IC50 value was around 150 µM (Fig. 1B).
The effects of capsaicin on mitochondrial cytochrome c release into cytoplasm and caspase-3 activity
The release of cytochrome c from the mitochondria was confirmed by measuring the absorbance of these molecules in cytosolic extracts after treatment of FaDu cells with 50, 100 and 200 µM capsaicin for 12 hours. A high level of released cytochrome c (about four-fold higher than untreated control) was detectable even at 50 µM capsaicin treatment (Fig. 2A). Caspase-3 activation was confirmed using a cellular caspase-3 enzyme activity assay. The activity of caspase 3 after capsaicin treatment increased in a time-dependent manner (Fig. 2B) and capsaicin-induced caspase 3 activity was blocked by pretreatment with Ac-DEVD-CHO (data not shown). These results indicate that capsaicin-induced apoptosis is mediated by cytochrome c triggered caspase 3 activation through the mitochondrial pathway. The increase in caspase 3 activity was further evidenced by detection of an increased level of inactivated PARP fragment (p85), a main substrate of executor caspases, by western blotting (Fig. 2C).
The effects of capsaicin on apoptosis related gene/protein expression
The expression of the Bcl-2 and Bax genes was analyzed by using RT-PCR. As shown in Fig. 3A, the expression of Bcl-2 was found to be decreased after treatment with capsaicin, whereas the expression of Bax was increased. The protein levels of apoptosis-associated proteins also followed a similar pattern, i.e. a decrease in Bcl-2 and an increase in Bad and Bax (Fig. 3B), indicating that the apoptotic response in FaDu cells is associated with the functions of mitochondria.
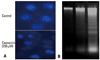
Capsaicin-induced DNA condensation and apoptosis in FaDu cells
To confirm that capsaicin-induced apoptosis is initiated via damage to cellular DNA, the effects of capsaicin on nuclear chromatin were investigated by staining the cells with DAPI. FaDu cells treated with 200 µM capsaicin for 24 hours showed condensed nuclei, seen by fluorescence microscopy (Fig. 4A). In addition, nuclear DNA fragmentation was additionally assessed by extracting nuclear DNA after treatment of the cells with 100 or 300 µM capsaicin for 24 hours. Capsaicin was found to induce internucleosomal degradation of DNA, resulting in ladder-shaped nucleosomal DNA fragments (Fig. 4B).
Flow cytometric analysis
Apoptotic cell death in FaDu cells was further confirmed by analyzing the sub-G1 DNA content using flow cytometry. Cells were treated with various concentrations of capsaicin for 24 hours, resuspended in propidium iodide dye, and subjected to flow cytometric analysis. As shown in Fig. 5, plots of the cell numbers versus propidium iodide intensity showed that the cell population undergoing apoptosis (in the sub-G1 peak) increased to 31.3% at 100 µM concentration of capsaicin and to 55.1% at 200 µM capsaicin, compared with 6.22% in untreated cells (Fig. 5).
Effects of capsaicin on cell cycle regulator proteins
To further evaluate the effects of capsaicin on cell cycle regulators such as cyclins and cyclin-dependent protein kinases (CDKs), FaDu cells were treated with/without 200 µM capsaicin for 24 hours. Capsaicin strongly suppressed the expression of G1/S regulators such as cdk-2, cdk-4 and cyclin D, indicating that capsaicin induces a cell cycle arrest at G1/S. The expression of cyclin B1 and cdk-1 showed a similar pattern, although the degree of inhibition was relatively weaker in this instance (Fig. 6).
DISCUSSION
Capsaicin, a spicy component of hot pepper, is widely consumed as a spice or food additive in Korea and other countries. Although there have been many previous reports of the tumor suppressive effects of capsaicin in human cancer cells, few studies have been undertaken to elucidate capsaicin-induced apoptosis in human oral cancer cells. In our current study, we demonstrated that capsaicin induces apoptosis in FaDu human pharyngeal squamous carcinoma cells via mitochondrial pathways. Capsaicin inhibits the growth and proliferation of FaDu cells in a dose- and time-dependent manners, and the apoptotic response was evidenced by characteristics such as cytochrome c release into cytosol, active caspase-3 generation, the expression of pro-apoptotic (or anti-apoptotic) markers, chromatin condensation in the cellular nucleus, and nuclear DNA fragmentation. The activity of caspase-3 was found to increase proportionally with the exposure time to capsaicin, suggesting that the cytochrome c release into the cytosol induced the apoptotic response by activating caspase-3, probably through an autocatalytic mechanism mediated by caspase-9.
The leakage of cytochrome c from the internal mitochondrial membrane into the cytosol initiates a "death wheel" involving active caspase-9.20-24 In our data, the cytochrome c levels in the cytosol of FaDu cells rapidly increased after exposure to a relatively low concentration of capsaicin (50 µM) and persisted even at high capsaicin doses, possibly due to cell death at these higher concentrations. An increase in caspase 3 activity was further supported by the detection of inactivated PARP fragment (p85), a principal substrate of executor caspases, on a western blot. The appearance of p85 fragment indicates that the DNA repair system in the cell has been damaged. Expression analysis further revealed a decrease in the anti-apoptotic Bcl-2 gene and an increase in the pro-apoptotic Bax gene. In addition, western blotting revealed a reduction of Bcl-2 with the enhanced expression of the pro-apoptotic Bax and Bad genes. Capsaicin-induced apoptotic cell death was further shown by the presence of nuclear condensation, DNA fragmentation and increased sub-G1 DNA content. Hence, our present findings confirm that capsaicin-induced apoptosis is mediated via mitochondrial-dependent pathways. Capsaicin-induced apoptosis was found to be mediated via the inhibition of cell cycle progression, since FaDu cells treated with capsaicin showed reduced expression of cyclin and cyclin-dependent protein kinases. Of particular note, the expression of all G1/S regulators such as cdk-2, cdk-4 and cyclin D, was found to be strongly affected by capsaicin, indicating that capsaicin induces a cell cycle arrest at G1/S, thereby suppressing cell cycle progression.
Previous studies of capsaicin-induced apoptosis in human cancer cells have revealed the involvement of various molecular mechanisms, depending on the tumor cell-types.16,17,19,25 However, effects of capsaicin on cell signaling mechanisms or on the molecular pathways of apoptosis in oral cancer cells are hardly known. To the best of our knowledge, the only report thus far is a recent study of apoptosis in SCC-4 human tongue cancer cells by Ip, et al.,19 exploring the molecular basis of the effects of capsaicin on apoptosis through mitochondria-dependent and -independent pathways. In this study, the apoptotic response in SCC-4 cells was associated with an increase in reactive oxygen species, Ca+2 generation and a disruption of the mitochondrial transmembrane potential. Our current results on the capsaicin-induced apoptosis in FaDu carcinoma cells are consistent with those of Ip, et al.19 in terms of the role of the mitochondrial pathway, although further investigations regarding the possible association of mitochondria-independent pathways remain to be conducted. Both studies clearly indicate that capsaicin has chemopreventive activities in cancer cells of different region of oral cavity, and thus has a great potential as a future therapeutic agent for oral cancer. The level of capsaicin (200 µM) in the present study is similar to the dose range of capsaicin used for the detection of non-neuronal cancer cells in other reports including SCC-4 cells.18 The physiological capsaicin dose for the oral cancer chemoprevention is unclear at this point and remains to be answered in future investigation.
In conclusion, capsaicin induces apoptosis in FaDu cells and this response is associated with mitochondrial pathways. The apoptosis may be mediated via a cell cycle arrest at the G1/S phase.





 XML Download
XML Download