

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Deficiency of 17α-hydroxylase is a rare autosomal recessive disorder which causes congenital adrenal hyperplasia.1 Defects in the synthesis of cortisol and compensatory hypersecretion of adrenocorticotropic hormone (ACTH) stimulate the synthesis of a large quantity of 11-deoxycorticosterone (DOC) and corticosterone in adrenal glands. High concentrations of DOC, which is a potent mineralocorticoid, lead to hyperaldosteronism which results in hypertension, hypokalemia, and a suppressed renin-angiotensin system. In gonads, the absence of 17α-hydroxylase activity prohibits the synthesis of androgens, causing pseudohermaphroditism in males and sexual infantilism with primary hypogonadism in females.2 Aldosterone excess with subsequent hypokalemia leads to the prolongation of the QT interval,3,4 which may increase the risk of fatal arrhythmias such as polymorphic ventricular tachycardia (Torsades de Pointes), although these are very rare in subjects with hyperaldosteronism. We report a case of male pseudohermaphroditism due to 17α-hydroxylase deficiency presented with malignant arrhythmias, Torsades de Pointes.
CASE REPORT
The 31-year-old woman was presented to our emergency room with an episode of resuscitated sudden cardiac arrest. She suddenly collapsed at her office, where, upon arrival, the emergency medical technician documented no pulse, and cardiopulmonary resuscitation was started and continued until the patient reached our hospital. An initial electrocardiogram revealed a normal sinus rhythm with left ventricular hypertrophy and a markedly prolonged QT interval (QTc=635 ms) (Fig. 1A). In the intensive care unit, recurrent episodes of Torsades de Pointes developed, which were terminated spontaneously (Fig. 1B) or by DC shock (Fig. 1C). Echocardiographic assessment revealed severe left ventricular hypertrophy with normal systolic function. The patient did have a past history of primary amenorrhea and had been treated for hypertension with hydrochlorothiazide and valsartan. Initial laboratory evaluation revealed severe hypokalemia of 1.6 mmol/L (reference, 3.5-5.5 mmol/L). Plasma renin activity was decreased (0.13 ng/mL/hr; reference, 0.15-2.23 ng/mL/hr), but aldosterone level was elevated (270.1 pg/dL; reference, 29.9-158.8 pg/dL). Plasma dehydroepiandrosterone (0.8 ng/mL; reference, 1.8-12.5 ng/mL) and cortisol (4.93 ng/mL; reference, 70-250 ng/mL) were decreased, while plasma 11-hydroxycorticosterone (76.0 ug/dL; reference, 8.0-30.0 ug/dL) and ACTH (420.5 pg/mL; reference, 7.2-63.3 pg/mL) were markedly elevated. Follicle stimulating hormone (30.9 mIU/mL; reference, 1.3-8.1 mIU/mL) and luteinizing hormone (19.6 mIU/mL; reference, 1.0-5.3 mIU/mL) were elevated; however, testosterone was undetectable.
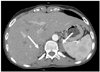
Abdominal computed tomography showed bilateral hyperplasia of adrenal glands (Fig. 2). Chromosome analysis for karyotype showed a 46, XY normal male karyotype, and genomic polymerase chain reaction direct sequencing for CYP17A1 confirmed the diagnosis of 17α-hydroxylase deficiency. After treatment with parenteral replacement of potassium and dexamethasone concurrent with antihypertensive medications, including captopril (300 mg/day), cilnidipine (20 mg/day), and spironolactone (100 mg/day), no more Torsades de Pointes was noted and QTc was normalized (Fig. 3).
DISCUSSION
We report a case of male pseudohermaphroditism with life-threatening arrhythmia as the first manifested symptom. A deficiency of 17α-hydroxylase is well known to cause male pseudohermaphroditism, combined with hyperaldosteronism. In this case report, hyperaldosteronism due to 17α-hydroxylase deficiency and diuretics might cause severe hypokalemia, leading to Torsades de Pointes. Low extracellular potassium produces hyperpolarization and prolongation of the action potential duration, resulting in Torsades de Pointes triggered activity.5 Moreover, a hypertrophic heart is known to be attributable to baseline increased transmural dispersion of repolarization and is more susceptible to develop Torsades de Pointes.6,7 Accordingly, this patient was at a very high risk of life-threatening arrhythmia and, surprisingly, her first presenting symptom was sudden cardiac arrest. Although the majority of cases of sudden cardiac arrest are caused by underlying cardiac disease vulnerable to fatal ventricular arrhythmia, including myocardial infarction with a low ejection fraction or congestive heart failure and electrical disorders,8 several endocrinologic disorders seem to present with sudden cardiac death. In the literature, fatal sudden cardiac arrest as a first manifested symptom was reported in patients with endocrinologic disorders such as anterior pituitary insufficiency and aldosterone producing adrenal adenoma.9,10 Although all of the endocrinologic disorders associated with sudden arrest show a similar mechanism of QT prolongation caused by hypokalemia, their underlying diseases are likely to be heterogeneous. Male pseudohermaphroditism is a rare endocrinologic disorder and sudden cardiac arrest is a very unusual presenting symptom of this disease. To our knowledge, this is the first report of male pseudohermaphroditism presented with sudden cardiac arrest. This case demonstrates that it is important to consider endocrinologic disorders among various causes in the diagnostic work-up, when a patient presents sudden cardiac arrest with a long QT interval.


 XML Download
XML Download