

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a fatal disease characterized by progressive degeneration of upper and lower motor neurons resulting in ventilatory failure and ultimately death within an average of 2-3 years of onset. It is the most common adult-onset disease with an incidence of two in 100,000 and a lifetime risk of one in 400-700. It has been commonly stated that familial amyotrophic lateral sclerosis represents 10% of ALS, of which 20% is associated with an autosomal dominant mutation in the Copper/Zinc superoxide dismutase 1 gene (SOD1).1 The remaining 90% of cases do not have an obvious family history of ALS and appear sporadically. Though the etiology of sporadic ALS (sALS) is unknown, epidemiological data indicate that sALS is a multifactorial disease in which unidentified modifying genetic factors contribute to pathogenesis.2 Many candidate genes have been implicated, but no single gene has been definitely shown to cause sALS until now.3 Humans have two nearly identical copies of SMN genes4: SMN1 located in the telomeric region on chromosome 5q13 and its centromeric copy, called SMN2. The full-length survival motor neuron (SMN) protein is encoded almost exclusively by SMN1. Conversely, the truncated SMN protein encoded by SMN2 lacks the exon 7 sequence and is less stable and more easily degraded. SMN is a ubiquitously expressed housekeeping protein that is essential for cell survival. Reduced levels of SMN cause harmful effects either to the splicing or to the axonal transport of a limited number of target genes that are specific to motor neurons.5 Homozygous deletion of SMN1 is present in nearly all cases of infantile spinal muscular atrophy (SMA). SMN2 is believed to modulate its phenotype.6
In this study, we determined SMN1 and SMN2 ratios in a small cohort of 25 Korean patients with sALS to investigate whether the SMN1 : SMN2 genotype is associated with susceptibility to and severity of sALS in a Korean population.
MATERIALS AND METHODS
Subjects
Twenty-five Korean patients were enrolled at the Neuromuscular Center of Gangnam Severance Hospital between 2003 and 2009. All of the patients were over the age of 15. They had both upper and lower motor neuron signs without any sensory symptoms. They were categorized as having definite, probable, or possible ALS according to El Escorial criteria.7 The following clinical data were obtained from the patients: age at disease onset, duration of disease until diagnosis, type of onset (spinal, bulbar), and El Escorial criteria. The clinical data were collected before the results of the genetic studies were available. A total of 100 Korean healthy individuals who had no family history of motor neuron disorder served as controls.8 We collected written informed consent and blood samples from all subjects. This study was approved by the Institutional Review Board of Yonsei University Health System and informed consent was obtained in accordance with the Declaration of Helsinki.
DNA analysis
DNA was extracted from the peripheral blood of each subject using the Easy-DNA kit (Invitrogen Life Technologies Corp., Carlsbad, CA, USA). A spectrophotometer was used to determine the concentration of the DNA samples. Using the commercially available SALSA MLPA KIT P021 SMA (MRC-Holland, Amsterdam, Netherlands), we genotyped the copy number of SMN1 and SMN2 in each subject. The kit contains a probe mixture for SMN1 exons 7 and 8, SMN2 exons 7 and 8, SMN (1 and 2) exons 1, 4, 6 and 8, NAIP (BIRC1), GTF2H2 (BTFFp44), RAD17, and CDH6 in the 5q13 region (16 probes) and several control fragments on different chromosomes (21 probes), as well as the standard multiple ligation-dependent probe amplification (MLPA) control probes used to determine ligation efficiency and DNA concentration. Details of probe sequences and gene loci are shown on the company's Web site (http://www.mrc-holland.com).
MLPA analyses were carried out according to the manufacturer's protocol (MRC Holland). In brief, 50-500 ng of DNA was denatured (98℃, 5 min) and hybridized with the probe set overnight at 60℃, using the SALSA probe mix. Ligation was performed using ligase-65 enzyme at 54℃ for 15 min. The reactions were inactivated by incubation at 98℃ for 5 min. PCR was performed using the specific SALSA PCR primers for 35 cycles (95℃ for 30 s; 60℃ for 30 s; 72℃ for 1 min) using GeneAmp® PCR System 9700 (Applied Biosystems Inc., Foster City, CA, USA). The PCR products were analyzed in a capillary DNA sequencer (ABI PRISM 310 Genetic Analyser, Applied Biosystems) with Genescan 3.7 Software (Applied Biosystems).
Data analysis was performed using the software GeneMaker® V 1.60 (SoftGentics, LLC, State College, PA, USA). The peak height of each specific probe was normalized by dividing it with the combined heights of the control probes. The relative peak height of each probe was compared with the relative peak height of the same probe in the control samples. A ratio of under 0.6 was taken as a sign of the presence of only one copy of the SMN1 or SMN2 gene.
Statistical analysis
We used SPSS statistical software (version 15, SPSS Inc., Chicago, IL, USA). Fisher's exact test was used to compare differences in the proportions of homozygous or heterozygous deletion of SMN between the patient and control groups. An ANOVA test was performed to compare differences in age of onset and medical research council (MRC) scale among the different patient genotypes. Significance was set at p<0.05.
RESULTS
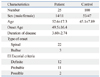
Table 1 shows the clinical characteristics of the patients according to sex, onset age, disease duration, type of onset and El Escorial criteria. In the control group, only sex and age were shown. There were 14 male and 11 female patients. The mean age was 52.6±17.3 years and the mean age of onset was 49.5±16.4 years. The mean disease duration before diagnosis was 3.60±2.74 years. Twenty-two patients had spinal onset and three had bulbar onset. According to El Escorial criteria, 12 patients were definite, 11 were probable, and two were possible ALS patients.
Table 2 shows SMN1 : SMN2 genotypes between patient and control groups. There was higher incidence of 2 : 0 genotype in patients with sALS (20% in sALS, 2% in control) (p<0.001). Calculation of total predicted SMN protein levels are displayed.
Table 3 shows onset age and initial motor grade in the patient group according to SMN1 : SMN2 genotypes. The onset age was younger in the 2 : 0 genotypes (34±15.38 years) than in the other genotypes (58±6.11, 52±14.62 and 45±7.07 years in 2 : 1, 2 : 2 and 2 : 3 genotypes) (p=0.049). The number of patients with an MRC scale higher than G4- was lower in the 2 : 0 genotype group (2, 5, 13 and 1 in 2 : 0, 2 : 1, 2 : 2 and 2 : 3 genotype) (p=0.02).
DISCUSSION
Two previous French studies6,9 showed an increased frequency of homozygous SMN2 deletions in groups with ALS. In this study, 20% of the Korean patients (5 out of 25) had homozygous deletions of SMN2 as compared to 2% (2 out of 100) of the healthy controls.
The relative predominances of male and spinal onset patients in our study were not significantly different from previous studies.1,4,10
The SMN protein is a well-documented component of the ribonuclear protein complexes required for mRNA transcription, splicing and trafficking.5 It also serves in local protein translation such as β-actin in the axons of spinal motor nerves and chaperone-like activity against mutant SOD1-mediated toxicity.5,11 SMN deficiency therefore exerts a harmful effect on neuronal cells and eventually causes development of motor neuron disease.
SMN protein expression can be simply estimated using the following formula4: SMN protein=SMN1 copy number+0.2×SMN2 copy number. Simple comparison of the total estimated SMN protein levels among genotypes suggests that low levels of SMN protein are associated with a higher risk of ALS (11 patients in SMN<2.4 versus 1 patient in SMN>2.4). However, the total level of SMN protein is not always associated with certain phenotypes. First, there are two normal control cases with heterozygous SMN1 deletions (1 : 1 and 1 : 3) that have SMN protein levels lower than 2.4. Second, more patients have the 2 : 2 genotype (13 cases, SMN protein 2.4) than have the 2 : 0 (5 cases, SMN protein 2.0) and 2 : 1 (6 cases, SMN protein 2.2). If disease severity and susceptibility correlate only with SMN proteins, the discrepancy of why the phenotypically normal control has heterozygous deletion of SMN1 and why more patients develop in the 2 : 2 genotype group cannot be explained. We hypothesize that small mutations such as deletions, splice mutations and missense mutations in SMN2 could lead to less SMN protein production. Eventually, this could indicate SMN2 as a risk factor for sALS.8,10 Third, there was one patient with the 2 : 3 genotype among the sALS patients. This suggests that a chromosome bearing two SMN copies may in fact be incapable of directing the synthesis of a normal quantity of or too much SMN protein.8 Another explanation is linkage disequilibrium with another ALS susceptibility gene mutation.8 Neuronal apoptosis inhibitor protein (NAIP) gene, at a distance of 15.5 kb from the SMN, or general transcription factor IIH, polypeptide 2 gene (GTF2H2) and small EDRK-rich factor 1A gene (SERF1A, formerly known as H4F5) are candidate genes.10 Larger deletions or duplications in that locus would influence SMN2.
In this study, we used onset age and initial MRC scale to measure disease severity among different SMN genotypes. Regardless of the limitations that have reported that onset age depends inevitably on the patient's memory, the 2 : 0 genotype group showed a significantly earlier onset age. Further objective disease severity scales to match genotypes should be identified in the future.
In conclusion, our data show that homozygous deletion of SMN2 could be one of the risk factors for sALS and correlates with disease severity expressed at a younger onset age and a lower MRC scale in the Korean population.


 XML Download
XML Download