

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The clinical manifestations of Behçet's disease were described by Hippocrates in the fifth century BC.1 However, in 1937, Turkish dermatologist Hûlusi Behçet was the first to comprehensively describe the symptom complex of recurrent oral aphthous ulcers, genital ulcers, and uveitis as a disease entity with the suggestion of a possible viral etiology.2
Although there are many synonyms for the condition, including triple symptom complex,3 syndrome de Behçet,4 Behçet's disease,5 Adamantiades-Behçet's syndrome,6 Behçet's multiple symptom complex,7 Behçet's syndrome, Adamantiades-Behçet's disease, mucocutaneous-ocular syndrome,8 mouth and genital ulcers with inflamed cartilage syndrome,9 and pseudo-Behçet's syndrome,10 Behçet's disease is the most commonly agreed upon terminology,11-13 and the representative international society on this disease uses the term Behçet's disease.
EPIDEMIOLOGY
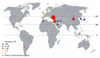
Behçet's disease exists worldwide although there are significant regional differences, with the highest number of incidences in the Mediterranean, the Middle East, and the Far East (Fig. 1, Table 1).10 The association of Behçet's disease with the ancient trading route known as the "Silk Road" and the distribution of HLA-B5 and its HLA-B*51 subtype provides important clues to its origin. Behçet's disease occurs most frequently between the latitudes 30° and 45° N in Eurasian populations.14-16 Although there are reports of Behçet's disease from other parts of the world including Mongolia, Russia, Brazil, Mexico, Columbia, Argentina, Chile, Cuba, Australia, and New Zealand, these reports total less than 200 patients.15,17 Behçet's disease may be an under-reported, HLA-B51 negative condition and is often life-threatening and/or associated with severe damage in native East African populations.18 Turkey demonstrates the highest prevalence of Behçet's disease in the world, with up to 421 per 100,000 persons affected. Iran, Israel, northern China, and Korea follow with the next highest prevalence. The countries with the lowest prevalence are the United Kingdom, Spain, Sweden, Portugal, and the United States, ranging from 0.3 to 6.4 per 100,000 persons.19-26 The numbers of patients with Behçet's disease registered by the National Health Insurance Corporation of Korea have been increasing. As of 2006, 15,554 patients with Behçet's disease (5,499 males and 10,055 females, 30.2/100,000 people) were registered with the Center for Genetic & Rare Diseases (Korea Centers for Disease Control and Prevention, Seoul, Korea).16
The onset of Behçet's disease typically occurs in the third or fourth decade of life, and it is rarely seen in children or patients above the age of 50. The clinical courses of childhood-onset Behçet's disease and late-onset Behçet's disease are relatively benign.16,21,24,27,28 The average time between the initial appearance of the first major symptom, usually oral ulcerations, and the development of a second major symptom in children is 8.8 years.27 The incidence of panuveitis decreases as age increases, while the incidence of anterior uveitis increases with age.28 Familial cases have been reported.29
Behçet's disease shows a male preponderance in Middle Eastern countries and the Mediterranean basin; however, women are more commonly affected in Japan and Korea.11,22,24,30 The mean age of onset for patients with the worst prognoses, such as those with ocular, gastrointestinal, neurologic, and vascular involvement, is significantly younger in males than in females.30 Behçet's disease was exacerbated in 67% of pregnant women and improved in 33%. In patients who experienced worsening of the disease during pregnancy, clinical exacerbation occurred most commonly during the first trimester, in about 78% of cases.31
ETIOPATHOGENESIS
The exact etiopathogenesis of Behçet's disease has not been clarified. However, many studies indicate that the disease may be triggered by environmental factors, such as infectious agents or pollution, in patients with backgrounds of genetic susceptibility (Fig. 2).25,32,33 Studies have shown that HLA-B*51 is associated with Behçet's disease, with more than 60% of patients with Behçet's disease testing positive for HLA-B*51.34 Recently, two reports of genome-wide association studies revealed that the MHC region of chromosome 6 was strongly associated with Behçet's disease, and HLA-B*51 is regarded as the primary association to Behçet's disease within the MHC region.34-36 In addition, meta-analyses identified that common variants of the IL10 and encoding interleukin 23 receptor (IL23R)-encoding interleukin 12 receptor beta (IL12B2) genes were strongly associated with Behçet's disease.35,36 IL23 is a proinflammatory cytokine that stimulates Th17 proliferation, increases the production of inflammatory cytokines, and increases the expression of IL-23 p19 mRNA in erythema nodosum-like skin lesions in patients with active Behçet's disease.34,37 IL10 is known as an anti-inflammatory cytokine that inhibits the action of proinflammatory cytokines, and the up-regulation of the CD4+ CD25+ T-regulatory cells in a Behçet's disease-like mouse model improved the inflammatory symptoms via IL10.38 Therefore, the IL10 and IL23R-IL12B2 genes may play major roles in the pathogenesis of Behçet's disease.
Autoimmune or autoinflammatory reactions in Behçet's disease are suggested to target primarily blood vessels, especially endothelial cells, causing the clinical presentation of vasculitis and/or thrombosis symptoms. Lee, et al.39 identified α-enolase as a target antigen of IgM-type anti-endothelial cell antibodies (AECA) in patients with Behçet's disease using proteomic techniques. Several mechanisms were proposed in order to explain the action of AECAs in the pathophysiology of inflammatory diseases, including the binding of AECA to endothelial cells resulting in cell activation, which may in turn increase secretions of chemoattractants and/or cytokines as well as secretion or inhibition of prostacyclin. AECAs might also trigger inflammatory processes by complement-dependent cytotoxicity and/or antibody-dependent cellular toxicity.
Infectious agents, especially herpes simplex virus (HSV) and Streptococcus sanguis (S. sanguis), have long been postulated as possible environmental triggers of Behçet's disease. Patients with Behçet's disease have significantly higher levels of S. sanguis in their oral flora than do healthy controls or patients with other diseases.33,40 Patients with Behçet's disease also show strong delayed cutaneous hypersensitivity reactions as well as oral aphthous ulcerations against streptococcal antigens after skin injection or oral prick tests with streptococcal antigens. Kaneko, et al.33 suggested that the Bes-1 gene and streptococcal 65 kDa heat shock protein from an uncommon serotype (KTH-1, strain BD113-20) of oral S. sanguinis are important extrinsic factors in the pathogenesis of Behçet's disease. Sera from patients with Behçet's disease, which react with recombinant human α-enolase, were revealed by proteomic techniques to cross-react with streptococcal α-enolase.40
HSV type 1 can be detected in saliva, intestinal ulcers, and genital ulcers by polymerase chain reaction in patients with Behçet's disease compared with healthy controls.41,42 In addition, a Behçet's disease-like mouse model was developed by inoculation of ICR mouse earlobes with HSV and demonstrated HSV DNA sequences in cutaneous and gastrointestinal ulcerative lesions.42 Although pretreatment or concurrent treatment with famciclovir in ICR mice could not prevent the development of Behçet's disease-like symptoms after HSV inoculation, famciclovir seemed to be effective in improving Behçet's disease-like symptoms and preventing recurrence in a symptomatic mouse model.43
Cytotoxic T-lymphocyte antigen-4 (CTLA-4) is a costimulatory molecule in T cells that plays a crucial role in immunoregulation, and reduced expression of CTLA-4 has been demonstrated in various autoimmune or autoinflammatory disorders.44 In patients with Behçet's disease, the role of serum soluble CTLA-4 levels and the associations between CTLA-4 polymorphisms and disease susceptibility have been reported.44 Recently, Sim, et al.44 reported that reduced expression of CTLA-4 in CD4+ T cells was observed in patients with active Behçet's disease after stimulation. In addition, decreased expression of CD86 and programmed death-ligand 1 were found in patients with active Behçet's disease.44
Behçet's disease has been described as both an autoimmune and an autoinflammatory disorder.45 Autoinflammatory diseases refer to inherited disorders characterized by episodes of recurrent inflammatory reactions of the innate immune system without remarkable provocation, especially by neutrophils; whereas autoimmune diseases present with significant levels of high-titer autoantibodies or antigen-specific T-cells.46 Infectious agents are suggested as triggering the inflammatory reaction mediated by the innate immune system, and "bacterial persistence" or autoantigen-activated antigen-presenting cells are required to sustain the inflammatory reaction mediated by the adaptive immune system.45
DIAGNOSIS AND CLINICAL MANIFESTATIONS
Because Behçet's disease has no pathognomonic laboratory findings or histological characteristics, diagnoses are made according to clinical criteria proposed by the International Study Group for Behçet's Disease47 or the Behçet's Disease Research Committee of Japan.48 Recurrent oral ulcers are the most frequently observed major symptom in Behçet's disease. However, it is difficult to differentiate between recurrent aphthous stomatitis (RAS) and recurrent oral ulcers in Behçet's disease.49 Patients with Behçet's disease are at a higher risk of major types of oral ulcers (deep and >10 mm in diameter) and a lower risk of minor types of ulcers (shallow and <10 mm in diameter) than are RAS patients.49 Patients with Behçet's disease more commonly have ulcers on two or more sites of the oral mucosa compared with RAS patients. Because minor symptoms, especially articular symptoms, are more commonly associated with Behçet's disease, RAS patients with additional major or minor symptoms should be monitored carefully for any progression to Behçet's disease.49
Diagnosing minor features that correlate with Behçet's disease is difficult because major manifestations emerge at different points in time throughout the course of the disease. Involvements of the gastrointestinal tract, vascular system, or central nervous system are closely correlated with mortality and morbidity, although life-threatening complications such as intestinal perforation, arterial occlusion, and aneurysm rupture are less frequent. However, the systemic manifestations of Behçet's disease lack reliable diagnostic criteria. Cheon, et al.50 reported novel diagnostic criteria for intestinal Behçet's disease in Korean patients with ileocolonic ulcers. Intestinal Behçet's disease was categorized into four groups: 1) definite (typical intestinal ulcers in colonoscopy plus complete, incomplete, or suspected types of Behçet's disease); 2) probable (typical intestinal ulcers in colonoscopy plus typical recurrent oral ulcers or atypical intestinal ulcers and complete, incomplete, or suspected types of Behçet's disease); 3) suspected (typical intestinal ulcers in colonoscopy without any extraintestinal symptoms or atypical intestinal ulcers plus typical recurrent oral ulcers); 4) and nondiagnostic (atypical intestinal ulcers without extraintestinal manifestations).50
18F-fluorodeoxyglucose positron emission tomography (FDG-PET) or PET/computed tomography (CT) is widely used for diagnosing diseases other than malignancies, including various causes of arthritis and vascular diseases. However, there have been few reports describing the clinical efficacy of using FDG-PET or PET/CT for diagnosing Behçet's disease.51,52 Denecke, et al.51 described the inflammatory activity of pulmonary artery aneurysms detected by FDG-PET/CT in patients with Behçet's disease. In addition, we reported eight patients with Behçet's disease with FDG uptake in cardiovascular lesions associated with Behçet's disease, including multiple pseudoaneurysms, aortitis, and arteritis associated with aortic regurgitation and aneurysmatic dilatation of the sinus of Valsalva, atherosclerotic changes of the proximal ascending aorta associated with aortic regurgitation, and multiple pulmonary artery aneurysms.52 Although these reports do not indicate that FDG-PET or PET/CT are necessary to diagnose patients with Behçet's disease, FDG-PET or PET/CT, which may be used to scan the entire body, may have clinical value as baseline workup tools against Behçet's disease if there is suspicion of associated malignancy or internal organ involvement.
Disease activity of Behçet's disease has been calculated clinically using the Behçet's Disease Current Activity Form 2006 (www.behcet.ws/pdf/BehçetsDiseaseActivityForm.pdf). However, because there are no novel laboratory markers that reflect disease activity in patients with Behçet's disease, serum erythrocyte sedimentation rate and C-reactive protein have been used to assess disease activity and clinical responses to treatment.53 In addition, S100A12, which contributes to the pathogenesis of Behçet's disease and is related to neutrophil hyperactivity, may reflect disease activity in Behçet's disease.54 Persistently high anti-streptolysin O (ASO) titers in patients with Behçet's disease indicate that streptococcal infections such as tonsillitis may be related to Behçet's disease symptoms, especially erythema nodosum-like skin lesions. In these patients, ASO titers can be used for the evaluation of disease activity, and antibiotic treatments might be effective for controlling the symptoms of Behçet's disease.55
TREATMENT
The treatment of Behçet's disease is symptomatic and empirical, but is generally specific to the clinical features of each patient.15,16,22,24,56-66 The appropriate management of Behçet's disease requires a multidisciplinary approach. Although various therapeutic modalities have been employed for Behçet's disease, the results of treatment are far from satisfactory. There is still no single effective drug for Behçet's disease in any clinical state. For evaluations of therapeutic efficacy, it is difficult to compare the results of different studies, and it is impossible to pool the results for calculating the effect sizes of treatment modalities due to the paucity of randomized controlled trials as well as the variability of the natural course of the disease and the limited number of cases available for clinical investigation.16
Despite limitations in the evaluation of therapeutic efficacy against Behçet's disease, treatment of Behçet's disease has become much more effective in recent years because of advances in disease pathogenesis and the increasing availability of a wide spectrum of therapeutic agents. Recently, a group of experts developed nine recommendations for the management of Behçet's disease by combining current evidence from controlled trials.59,60 The European League Against Rheumatism recommendations for the management of Behçet's disease suggests that recommendations related to the eye, skin-mucosa disease and arthritis are mainly evidence based, but that recommendations on vascular disease, neurological and gastrointestinal involvement are based largely on expert opinion and the results of uncontrolled open trials and observational studies.59 Corticosteroids are commonly used to treat clinical manifestations of Behçet's disease as a monotherapy or in combination with immunosuppressants. Although they have beneficial effects against acute inflammation, no definite evidence has indicated that they are effective for controlling progression of the disease, and the adverse effects of long-term use must always be considered.
Tumor necrosis factor (TNF)-blocking agents such as infliximab, etanercept, and adalimumab have been reported to have some success in patients with Behçet's disease who have mucocutaneous or gastrointestinal lesions, neurological disease, or even pulmonary aneurysms.22,67 There is enough published experience to suggest that TNF blockade represents an important therapeutic advance for patients with severe disease who are resistant to standard immunosuppressive regimens and for those patients with contraindications or intolerance to these treatments.67 However, the high cost, need for injections, troublesome toxic side effects, and lack of long term evidence for therapeutic efficacy are the main limitations to the widespread acceptance of anti-TNF-α agents as a first-line choice for Behçet's disease management. The adverse effects of anti-TNF-α agents include infection, autoimmune reactions, lymphoproliferative disorders, delayed hypersensitivity reactions, as well as neurologic, cardiac, and gastrointestinal symptoms. In addition, cases of relapse and new onset of uveitis during anti-TNF-α treatments have been reported. The mechanism of new onset uveitis is not well known thus far.68,69
Nowadays, there are some optimistic viewpoints about managing Behçet's disease because researches to reveal the pathogenesis and to develop new therapeutic agents are actively being held. In an effort, immune tolerance utilizing the peptides of Hsp-65/60 has recently been considered as a possible new therapy for patients with Behçet's disease.70 The fact that the most typical cases of Behçet's disease, those with all of the 4 major symptoms, have been decreasing in Japan, although the reason remains uncertain, may be another relatively good news.71
Therapeutic agents should be selected after thorough evaluations of immune status through a variety of tests and after determining any aggravating or provoking factors. Combination regimens are generally accepted to be more effective than single agent regimens. The most effective management involves early diagnosis and clinical intervention with continuous follow-up. Consequently, it may be possible to reduce the risk of serious complications and socioeconomic costs due to Behçet's disease. The therapeutic approach to Behçet's disease is summarized in Fig. 3. However, further studies are still necessary to find the most appropriate therapy and diagnostic markers to overcome this rare, intractable disease, which often leads to blindness and fatal systemic involvement.


 XML Download
XML Download