

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Autophagy is a part of cellular system involved in maintaining homeostasis by degrading long-lived cellular constituents.1 It also plays critical roles in providing nutrients under starvation and neonatal periods.2,3 There are three types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy.4 In chaperone-mediated autophagy, signaling motif containing molecules are transported with the chaperone HSC70 via LAMP-2A into lysosomes.5-8 In contrast to microautophagy, which is characterized by the removal of constituents via budding of an autophagic body at the lysosomal membrane, macroautophagy forms a double-layered membrane vesicle, called an autophagosome. The autophagosome is formed via the elongation of a cup-shaped membrane, and two ubiquitin-like conjugation systems are involved in autophagosome propagation.9 At least 30 genes, termed autophagy-related genes (Atg), regulate the process of autophagy in yeast.10 Once formed, the outer membrane of the autophagosome fuses with a lysosome, where cellular contents are degraded within by lysosomal hydrolase and recycled.11
Beyond maintaining homeostasis, autophagy is involved in multiple biological processes including development, aging, and degeneration.12 Not surprisingly, aberrant regulation of autophagy induces many diseases such as cancer, neurodegenerative disease, and myopathies.13,14 Autophagy also has diverse functions in immunity. Various intracellular bacteria, viruses, and protozoans are removed from host cells by autophagy, and endogenous antigens are processed and presented to major histocompatibility complex (MHC) class II via autophagy.15-21 In this review, we focus on the role of autophagy in innate recognition of pathogens and adaptive immune responses.
AUTOPHAGY IN PATHOGEN RECOGNITION
Autophagy in TLR signaling
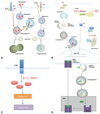
In response to pathogens, various types of pattern recognition receptors (PRRs) recognize pathogen-associated molecular patterns (PAMPs) and mediate signals to defend to pathogens.22 Among PRRs, Toll-like receptors (TLRs) respond to lipopolysaccharides (LPS), lipotechoic acid and flagellin on cell surface membranes, as well as to viral/bacterial nucleic acids on endosomal membranes.23 TLR4, a receptor for bacterial LPS, triggers both MyD88- and TIR domain-containing adapter-inducing interferon-β (TRIF)-dependent signaling pathways. The IKK-α-IKK-β-NEMO complex and TBK1-IKKi complex mediate the activation of the transcription factors NF-κB and interferon regulatory factor 3 (IRF3), respectively. In turn, they induce the transcription of proinflammatory cytokines and type I interferons (IFNs).24,25 TLR4 signaling via the TRIF-p38 axis, but not via MyD88, induces the formation of an autophagosome for the elimination of Mycobacteria bacilli.26 Atg6 and Beclin-1 are required in this process (Fig. 1A). Interestingly, in autophagy-deficient cells, IL-1β and IL-18 production is enhanced in response to LPS.27 Macrophages lacking Atg16L1 induce high-levels of reactive oxygen species (ROS), which in turn activates caspase-1, leading to the processing of IL-1β. However, in macrophages of wild-type mice, the generation of ROS is inhibited by autophagy-related proteins, and in turn, limited amounts of IL-1β are produced (Fig. 1C).
In addition to TLR4 signaling, other TLRs also activate autophagy machinery to eliminate pathogens. TLR7 signaling, induced by two different ligands, single-stranded RNA and imiquimod, induces the formation of autophagosomes, characterized by microtubule-associated light chain 3-green fluorescent protein (LC3-GFP) puncta formation for the elimination of Bacillus Calmette-Guerin.28,29 The induction of autophagy is dependent on MyD88. Here, both Atg5 and Beclin are required for the induction of autophagy in macrophages after stimulation of TLR7 (Fig. 1A).
Although most TLR signaling induces autophagosome formation, some link autophagy with the enhancement of phagocytosis. When zymosan (a particle of fungal cell walls) is phagocytosed, LC3, an autophagosome marker, is rapidly recruited to the phagosomal membrane.30 This process depends on Atg5 and Atg7. Although translocation of LC3 to the phagosome is not associated with autophagosome formation, it promotes the phagosome to fuse with lysosomes in murine macrophages. Interestingly, recruitment of LC3 to phagosome depends on TLR2, but not on MyD88 (Fig. 1A).
In addition to the formation of autophagosomes and maturation of phagosomes by autophagy-related proteins to promote TLR-mediated pathogen elimination, autophagy supports delivery of viral nucleic acids to lysosomes where TLR recognition occurs.31 Within the TLR family, TLR3, TLR7, TLR8 and TLR9, located in the endosomal compartment, sense endocytosed viral nucleic acids.32 In the case of vesicular stomatitis virus (VSV), plasmacytoid dendritic cells (pDCs) recognize replication intermediates in the cytosol, rather than single-stranded viral RNA via TLR7 in the endosome.31 In recognition of cytosolic viral PAMP by TLR7, autophagy mediates the transport thereof into lysosomes (Fig. 1B). Consistently, pDCs lacking Atg5 failed to secrete IFN-α and IL-12p40 in response to VSV infection, in a previous study. In addition to VSV, a TLR9 ligand herpes simplex virus (HSV)-1 failed to induce IFN-α production in Atg5-deficient pDCs, while the IL-12 response remained intact in these cells.31 These data suggest that the IFN-α signaling pathway is impaired in the absence of Atg5. However, the precise nature of the differential control of NF-κB versus IFN-α induction pathways in pDCs by autophagy remains to be determined.33
Autophagy in NLR signaling
Although TLRs are the most well-known PRRs, some bacterial structures are recognized by intracellular sensors, called nucleotide-binding oligomerization domain (NOD)-like receptors (NLRs).34,35 NLRs are a family of cytoplasmic molecules that recognize bacterial cell wall components, such as peptidoglycans, and initiate signaling cascades to activate inflammatory responses.35 NLRs comprise two different subsets, NODs and NALP (NACHT-, LRR- and pyrin domain-containing proteins). When NLRs recognize bacterial peptidoglycans, they initiate signaling pathways by recruiting protein kinase, which in turn activates NF-κB and AP-1, leading to production of cytokines and other molecules of innate immunity.36 Recent studies have shown that stimulation of NOD2 by muramyl dipeptide induces activation of autophagy and clearance of bacteria.37 This effect depends on autophagy-related proteins, such as Atg5, Atg7, and Atg16L1, as well as receptor interacting serine-threonine kinase-2, a regulator of the NOD2 signaling pathway. When NOD2 is stimulated with bacterial components, the autophagy protein Atg16L1 is recruited to the plasma membrane at the bacterial entry site.38 This then promotes bacterial trafficking to the autophagosome, maturation of the autophagosome via fusion with a lysosome, and facilitates antigen loading to MHC class II molecules to enhance antigen presentation to CD4 T cells. As expected, DCs isolated from patients of Crohn's disease, expressing risk alleles for NOD2 or Atg16L1, are not able to induce autophagy pathways, and in turn, fail to promote bacterial clearance and antigen presentation.37,38
AUTOPHAGY IN CYTOSOLIC PRRS
Most cell types utilize cytoplasmic receptors, RIG-I-like receptors (RLRs), to bind to viral RNA. These cytosolic sensors are caspase recruitment domain (CARD)-containing proteins, consisted of retinoic acid inducible gene I (RIG-I) and melanoma differentiation associated gene 5.39-41 Via their CARD domain, RLRs signal with IFN-β promoter stimulator-1 (IPS-1) and activate transcription factors, including IRF3 and NF-κB, to produce type I IFN and proinflammatory cytokines, respectively.36 Although autophagy-related proteins such as Atg5 mediate transport of viral replication intermediates from the cytosol to lysosomes, where recognition thereof by TLR7 occurrs,31 autophagy-related proteins engaged in RLR-mediated viral sensing act as negative regulators of anti-viral responses.42 The Atg5-Agt12 conjugate directly associates with CARD domains of RIG-I and IPS-1 to suppress type I IFN production (Fig. 1B). Consequently in Atg5-deficient mouse embryonic fibroblasts (MEFs), RLR signaling resulted in the overproduction of type I IFNs and resistance to VSV infection.42 Moreover, the accumulation of dysfunctional mitochondria and ROSs, associated with dysfunctional mitochondria in Atg5-deficient cells, potentiated RLR signaling in response to viral infections.43
In cases where double-stranded DNA (dsDNA), derived from bacteria or DNA viruses, is detected, the sensing and regulation mechanisms thereof are still unclear. Recently, studies have shown that dsDNA can induce the production of type I IFNs.44,45 In this process, the translocation and assembly of stimulator of IFN genes (STING) and TANK-binding kinase 1 (TBK1) are required.46-49 After sensing dsDNA, STING, a multispanning membrane protein, is translocated from the ER to the Golgi apparatus and assembled with TBK1, which phosphorylates the transcription factor IRF3, resulting in the production of type I IFN. Atg9a, an essential component of autophagy, is co-localized with STING in the Golgi apparatus and controls the dynamic translocation of STING and assembly with TBK1 (Fig. 1B).50 In Atg9a-deficient MEFs, but not in Atg7-and Atg16L1-deficient MEFs, the translocation of STING from the Golgi apparatus to the cytoplasmic punctate structures and assembly with TBK1 are greatly enhanced, which, in turn, induces aberrant activation of type I IFN responses.25 Thus, these results demonstrated that Atg9a plays a role in the fine-tuning of innate immune responses.
AUTOPHAGY IN ANTIGEN PRESENTATION
Autophagy has been known to be involved in innate immunity by directly defending against pathogens and by promoting innate recognition of pathogens. Furthermore, autophagy facilitates adaptive immune responses such as antigen presentation.51,52 Generally, intracellular cytosolic or nuclear antigens are presented by MHC class I molecules to CD8 T cells after proteosomal hydrolysis, whereas extracellular antigens are presented by MHC class II molecules to CD4 T cells after lysosomal degradation.53,54 Thus, it is somewhat expectable that lysosomal degradation of cytoplasmic molecules by autophagy can provide antigens presented by MHC class II molecules to activate CD4 T cells (Fig. 1D). Indeed, cytosolic and nuclear antigens are composed of 20-30% of natural ligands of MHC class II molecules.55-57 Recent studies revealed that autophagy is involved in the MHC class II processing and presentation of various intracellular antigens to CD4 T cells.17-21 Macrophages, which are unable to present several epitopes of endogenous C5 protein (5th component of mouse complement) with MHC class II molecules, became able to do so when they were treated with low doses of the lysosomotropic agent ammonium chloride.17 Also, MHC class II presentation was abrogated when treated with inhibitors of the class III PI3 kinase, which is known to inhibit autophagy induction. The presentation of peptides from the tumor antigen mucin 1 with transfection into DCs by RNA electroporation was inhibited to present by treatment of the inhibitor of class III PI3 kinase, 3-methyladenine (3-MA) and Wortmannin.18 Endogenous presentation of cytosolic protein neomycin phosphotransferase II (NeoR) on MHC class II was mediated by autophagy. NeoR was sequestrated into autophagosomes and delivered to lysosomes. This pathway was blocked by 3-MA and Wortmannin.19 Another report indicated that the endogenous nuclear antigen 1 of EBV (EBNA1) gains access to the MHC class II presentation pathway via autophagy. EBNA1 was accumulated in autophagosomes when lysosomal acidification was inhibited. Further, intracellular EBNA1 processing for MHC class II presentation was inhibited after siRNA-mediated silencing of Atg12, an essential protein involved in autophagosome formation.20 A more recent report showed that the targeting of a model antigen, influenza matrix protein 1 (MP1), to autophagosomes enhanced MHC class II presentation.21 Via fusion of MP1 to LC3, an autophagosome-associated protein, antigen presentation with MHC class II to MP1-specific CD4 T cells was enhanced 20-fold, while MHC class I presentation to CD8 T cells was not affected. Furthermore, siRNA-mediated silencing of Atg12 inhibited MP1-LC3 transport to the MHC class II compartment. This result implicated that autophagy plays important roles in delivering cytosolic antigens to the MHC class II compartment for enhanced MHC class II presentation to CD4 T cells.
In recent years, studies have revealed that autophagy promotes MHC class II presentation of extracellular antigens to CD4 T cells.58,59 In mice with dendritic cell (DC)-conditional deletion in Atg5, CD4 T cell priming after herpes simplex virus infection was impaired.58 Atg5-deficient DCs showed a pronounced defect in the fusion of phagosomes with lysosomes and consequent processing and presentation of phagocytosed antigens containing TLR stimuli for MHC class II. Similarly, autophagy mediated engulfment of endocytosed extracellular human immunodeficiency virus-1 (HIV-1) and promoted the processing of peptides for presentation to MHC class II loading compartments.59 In this report, envelope proteins of HIV-1 downregulated autophagy pathways by enhancing mammalian target of rapamycin signaling, and consequently autophagy failed to promote MHC class II presentation.
AUTOPHAGY IN THERAPY OF HUMAN DISEASE
The involvement of autophagy in immunity suggests that autophagy may be a target for therapy in various human diseases. For example, antigens from pathogens, which induce severe infectious diseases and are targeted by autophagy pathways, can be used to develop vaccines in order to enhance MHC class II presentation to CD4 T cells. Also, autophagy may be developed to antimicrobial agents based on the ideas that enhancing or inhibiting autophagy affects host resistance to viral, bacterial and parasitic infection. Furthermore, as mutations of autophagy genes such as Atg16L1 are known to be causal factors of Crohn's disease,37,38 modulation of autophagy might ameliorate Crohn's disease, if it can compensate for the functions of Atg16L1. The basic function of autophagy, degrading damaged or long-lived organelles might be a useful target for cancer therapy. However, controversy remains on whether enhancing or inhibiting autophagy might have beneficial effects on cancer therapy.60-63 In addition, imbalanced control of autophagy has demonstrated adverse effects including disturbed negative selection with consequent autoimmunity.64,65 Therefore, further comprehensive understanding of autophagy will be needed for proper modulation thereof without unwanted immunologic consequences.
CONCLUDING REMARKS
Recent studies have demonstrated that autophagy is important to immune responses. With its most notable function of degrading cytosolic constituents, it has also been shown to degrade intracellular microbes. In addition to directly defending against pathogens, autophagy facilitates the sensing of pathogens by pattern recognition receptors and modulates consequent signaling pathways in multiple ways. Autophagy promotes the elimination of some bacteria by autophagosome formation, whereas some fungal infections are resolved through maturation of phagosomes with the assistance of autophagy. Moreover, autophagy promotes the audetection of viral infections by delivering the cytosolic replication complex of the virus to the site of endosomal sensor recognition. Although autophagy usually enhances the signaling pathway after PRRs sense the pathogens, it can also negatively regulate the signaling pathway in certain cases. In addition to innate recognition, autophagy can be used to facilitate MHC class II presentation of endogenous intracellular antigens as well as phagocytosed or endocytosed extracellular antigens to activate CD4 T cells. Since autophagy plays multiple roles in immunity, autophagy is a potential target for therapies in various human diseases. However, it is difficult to determine whether induction or suppression of autophagy has beneficial effects, since autophagy is involved in multiple pathways of immune regulation. Thus, further comprehensive understanding of autophagy will provide a more integrated picture of how this system controls innate and adaptive immunity.
 XML Download
XML Download