

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Acetaminophen (APAP, Tylenol®, also known as paracetamol, N-acetyl-p-aminophenol) has analgesic and antipyretic properties and is considered safe in therapeutic doses. However, overdose of APAP can lead to severe liver injury in both humans and experimental animals. Investigations into the mechanisms underlying APAP toxicity have revealed metabolic conversion of APAP to a highly reactive intermediate, N-acetyl-p-benzoquinonimine (NAPQI),1 by cytochrome p450s, which are abundant enzymes in liver cells. The active NAPQI metabolite can be detoxified by conjugation with glutathione (GSH), a non-protein thiol with both oxidant-scavenging and redox-regulating capacities. Following overdose with APAP, NAPQI may be generated at much higher levels than can be detoxified by conjugation with available GSH. As cellular pools of GSH become depleted, NAPQI freely binds to functional proteins within the cells, leading to hepatotoxicity. Metabolism of APAP is also accompanied by the formation of reactive oxygen species (ROS), which initiate lipid peroxidation. This peroxidation is a critical factor that causes destruction and damage to cell membranes and finally leads to necrosis or apoptosis of the liver cells.2
The number of intoxications by APAP is worrisome.3 In 2006 alone, the American Association of Poison Control Centers implicated acetaminophen in nearly 140,000 poisoning cases, in which more than 100 patients died.4 Its incidence has been steadily increasing over the past decade, and acetaminophen is now the most common cause of acute liver failure.
Toxicity studies of APAP are now expanding to the molecular level with the use of microarray techniques to depict gene expression profiles in the liver. Toxicogenomics is a new discipline of molecular toxicology that incorporates the emerging technologies of genomics and bioinformatics for the quantitation and characterization of changes in gene expression in response to exposure to toxicants. Gene expression profiling in several laboratories has also allowed for prediction of subtle injuries to the liver induced by APAP. Functional classification of the differentially expressed genes identified those associated with stress responses, the cell cycle, growth inhibition, cell death, structural components, cell signaling, and inflammation.5,6
However, few studies have been conducted on cardiac toxicity induced by APAP. Myocardial damage with subendocardial hemorrhage and focal necrosis was reported following an APAP overdose.7,8 Details on the cardiotoxicity caused by APAP have never been further investigated, and little is known regarding the precise molecular events occurring within the myocardium following APAP insult. Although the cytochrome p450 enzymes are most abundant in the liver, some studies on metabolic enzymes in the heart have recently indicated the presence of cytchrome p450s,9,10 so the possibility of metabolic activation of APAP in the heart cannot be excluded.
The objectives of the present study were to identify APAP-induced alterations in gene expression in the heart and to determine whether alterations that occur in heart tissue differ from those known to occur in the liver following administration of a toxic dose of acetaminophen. Deregulation of normal gene expression by APAP may be indicative of potential adverse effects in the absence of the occurrence of overt toxicity. For this study, microarray technology was used for heart gene profiling as it provides a rapid and efficient method for large-scale profiling of gene expression changes.
MATERIALS AND METHODS
Chemicals and animals
APAP, Tween 80, and carboxymethyl cellulose (CMC) were purchased from Sigma-Aldrich (St. Louis, MO, USA). APAP was dissolved in a 0.5% (w/v) CMC solution containing 0.1% Tween 80. Male Sprague-Dawley rats, 6 weeks of age, were obtained from Orient Co. Ltd (Seongnam, Korea). The animals were acclimatized to laboratory conditions for one week, and maintained on a 12 h light/dark cycle in the animal room at 23±1℃ and humidity 50±5%. They were provided with food and drinking water ad libitum. APAP was orally administered to the rats as a single dose of 1,000 mg/kg. The dose level was set at the maximum tolerated dose level, based on clear signs of toxicity with little or no lethality.11 The respective vehicle control was administered to the non-treated control group through the same oral route.
Serum biochemistry and hematology
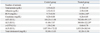
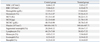
Sera were prepared from whole blood obtained from the treated group and the non-treated control group at 24 hours after APAP treatment. Clinical chemistry analysis was performed using an automated clinical chemistry analyzer (Hitachi7180, Hitachi, Japan). Analysis included the total protein, albumin, blood urea nitrogen (BUN), creatinine, total cholesterol, and the activities of marker enzymes such as aspartate aminotransferase (AST), alanine aminotransferase (ALT), γ-glutamyl transpeptidase (γ-GTP), and alkaline phosphatase (ALP). Hematology analysis was performed on whole blood collected using ethylenediaminetetraacetic acid (EDTA) as an anticoagulant. Erythrocyte, platelet, and leukocyte counts, hematocrit and hemoglobin concentrations, mean cell volumes (MCV), mean cell hemoglobin (MCH), and mean cell hemoglobin concentrations (MCHC) were determined using an automated hematology analyzer (HemaVet850, Drew-Scientific Group, San Diego, CA, USA).
Histopathology
Livers and hearts were removed and prefixed in 10% (v/v) neutral buffered formaldehyde for histopathological examination. The fixed heart and liver tissues were trimmed, dehydrated, embedded in paraffin, sectioned, mounted on glass slides, stained with hematoxylin and eosin, and examined by light microscopy. A histopathologist evaluated all heart and liver sections in a blinded fashion and recorded observations.
RNA isolation and gene expression profiling
Gene expression analyses were performed using Affymetrix GeneChip® Rat Gene 1.0 ST oligonucleotide arrays. The sample preparation was performed according to the instructions and recommendations provided by the manufacturer. The treated rats that showed definite histopathological abnormalities in the liver but not in the heart by conventional toxicology were selected for comparison with non-treated rats. Small pieces of tissue from three rats were pooled and RNA was extracted. In brief, the total RNA was isolated using RNeasy Mini Kit columns as described by the manufacturer (Qiagen, Hilden, Germany). RNA quality was assessed with an Agilent 2100 bioanalyzer using the RNA 6000 Nano Chip (Agilent Technologies, Amstelveen, The Netherlands), and RNA quantity was determined using an ND-1000 Spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA). The 260/280 nm ratio of total RNA used in the study was 1.8-2.0. For each RNA sample, 300 ng of total RNA was converted to double-stranded cDNA, as recommended by the manufacturer. Using a random hexamer incorporating a T7 promoter, amplified RNA (cRNA) was generated from the double-stranded cDNA template though an in-vitro transcription reaction and purified with the Affymetrix sample cleanup module. The corresponding cDNA was regenerated through a random-primed reverse transcription using a deoxyribonucleotide triphosphate (dNTP) mix containing deoxyuridine triphosphate (dUTP). The cDNA was then fragmented by uracil DNA glycosylase (UDG) and apurinic/apyrimidinic (AP) endonuclease 1 (APE 1) restriction endonucleases and end-labeled by terminal transferase reaction incorporating a biotinylated dideoxynucleotide. Fragmented end-labeled cDNA was hybridized to the GeneChip® rat Gene 1.0 ST arrays for 16 hours at 45℃ and 60 rpm, as described in the Gene Chip Whole Transcript Sense Target Labeling Assay Manual (Affymetrix). After hybridization, the chips were stained and washed in a Genechip Fluidics Station 450 (Affymetrix) and scanned with a Genechip Array scanner 3000 7G (Affymetrix). Affymetrix data were extracted, normalized, and summarized using the robust multi-average method implemented in the Affymetrix Expression Console. Highly expressed genes that showed a 2-fold change in expression were selected. The results were classified using hierarchical and K-mean clustering algorithms implemented in TMEV software 4.0. Differentially expressed genes were then classified based on their known biological functions using the Database for Annotation, Visualization, and Integrated Discovery software. Functional classification was based on both Gene ontology and KEGG pathway analysis.
RESULTS
Serum biochemistry, hematology, and histopathology
Clinical observations and neurobehavioral assessment showed no treatment-related changes for 24 hours after a high dose (1,000 mg/kg-bw) of APAP. Body weight and consumption of water and food did not change significantly after treatment with APAP. The toxic effects of APAP were evaluated by measuring serum levels of tissue-specific enzymes and by histopathological examination. In APAP-treated rats, significant increases in ALT and AST activity were noted 24 hours after treatment of APAP. Changes in enzyme activity in serum and blood cells after administration of APAP are shown in Table 1 and 2. Serum levels of ALT were 300.80±322.29 IU/L (mean±SD) in the treated group (n=5) and 41.00±7.07 IU/L in the controls (n=4). AST levels were 702.80±491.11 IU/L in the treated group (n=5) and 104.25±11.08 IU/L in the controls (n=4). BUN, an indicator of kidney damage, was also increased to 14.00±2.44 mg/dL in the treated group; the control group level was 9.75±0.50 mg/dL. Other parameters of serum biochemistry, including total protein, albumin, total cholesterol, creatinine, γ-GTP, and ALP were not changed by APAP administration (Table 1). Conventional analysis revealed no changes in hematological parameters including white blood cells, red blood cells, hemoglobin, hematocrit, MCV, MCH, MCHC, platelets, segmented neutrophils, lymphocytes, monocytes, eosinophils, and basophils (Table 2).
Histopathological changes observed in livers from APAP-treated rats consisted of minimal to moderate degrees of centrilobular hepatocellular necrosis and inflammatory cell infiltration, which supported the results of serum transaminases. The arrows in Fig. 1 indicate the abnormal regions of necrosis in liver. However, no histopathological alterations were observed in the hearts of the APAP-treated rats in the study.
Gene expression profiling
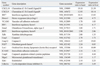
Using microarrays, the expression levels of more than 20,000 genes were analyzed in heart and liver tissues. Twenty-four hours after treatment with a single dose of 1,000 mg/kg of APAP by oral gavage, 271 genes were up-regulated and 81 genes were down-regulated in the heart, while 551 genes were up-regulated and 404 genes were down-regulated in the liver, based on a two-fold criterion.
Of the 271 up-regulated genes in the heart, 150 gene counterparts were also up-regulated in the liver. However, 121 genes were uniquely up-regulated in the heart, while 401 genes were up-regulated only in the liver. Among the 81 genes down-regulated in the heart, 13 genes were also down-regulated in the liver, while the remaining 68 were uniquely down-regulated in the heart. Among the 404 genes down-regulated in the liver, 391 were uniquely down-regulated in the liver and not in the heart (Table 3). The induction levels of some genes in the heart were significantly higher than those in the liver, while other genes showed the opposite.
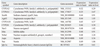
A partial list of the genes induced in the heart and their expression ratios in the heart and liver is shown in Table 4. In the liver, the expression of genes related to inflammation, oxidative stress, and apoptosis were induced, as reported previously.12,13 Many of the genes induced in the liver were also induced in significant levels in the heart. For example, CXCL10, a chemokine ligand 10 gene, was up-regulated 19.2-fold in the heart and 21.6-fold in the liver. Similarly, expression of interferon (IFN) regulatory factor 7 (IRF-7) was increased 10.1-fold in the heart and 13.8-fold in the liver. Heme oxygenase 1 (Hmox1), a well-known molecular biomarker of oxidative stress, was increased 6.1-fold in the heart and 4.8-fold in the liver. The expression of caspase (Casp) enzymes, which are involved in cellular apoptosis, was significantly increased both in the heart and the liver. As mentioned, the results of conventional toxicity endpoints, including serum biochemistry and histopathology, showed some abnormalities only in the liver, but not in the heart (Table 1, 2 and Fig. 1). However, the expression of many genes related to oxidative stress, inflammation, and apoptosis were altered in the heart as well as in the liver. This suggested the possibility of intoxication in the heart based on molecular level standards. The partial list of genes repressed in the heart and their expression ratios in the heart and liver are shown in Table 5. In the heart, many cytochrome p450 enzymes were repressed. The CYP3A group of cytochrome p450 proteins, a well-known group of enzymes that metabolize drugs, including APAP, was significantly repressed in both the heart and liver. Other groups of cytochrome p450s were also repressed by APAP. Among the repressed genes in the heart, the identity and function of many of the revealed genes are still unknown (data not shown).
DISCUSSION
Drug-induced cardiotoxicity is emerging as an important issue following anticancer treatments. The symptoms encompass a number of heterogeneous side effects including arrhythmias, changes in blood pressure, myocardial ischemia, thrombosis, and impairment in myocardial contraction and/or relaxation. In the past, the risk of heart complications was less evident because life span after chemotherapy was too short for the cardiovascular effects to be of major concern. With the progress in cancer therapy, however, cardiotoxicity has now become a pivotal issue. A number of targeted drugs in addition to conventional anthracyclines now represent the current mainstay of treatment for several forms of cancer, but their use is hampered by cardiac side effects. Many patients with neoplasms who undergo cancer therapy are now at a substantial risk for deterioration of their cardiovascular health. However, the cardiotoxic mechanisms of anticancer drugs have not yet been well studied. Oxidative stress seems to be the common pathway leading to mitochondrial damage, which then progresses to DNA damage, ATP blockade, cellular apoptosis, and finally cardiac dysfunction.14
Many other drugs in addition to anticancer drugs, such as APAP, are known to induce oxidative stress and to cause side effects during their metabolism in the liver.1,2 However, no similar studies have yet been conducted on the cardiotoxicity of APAP. Although APAP is considered safe at therapeutic doses, it may generate oxidative stress in the heart as well as the liver, thereby causing adverse cardiac side effects. In the absence of significant toxicity by conventional toxicity end-points, changes in gene expression are critical for monitoring biological effects in the heart, and these changes may predict possible cardiotoxicity in response to APAP administration.
In Fig. 1, serious liver damage was observed, based on histopathological criteria, when rats were orally treated with APAP 1,000 mg/kg-bw, but no histopathological alterations were observed in the heart. However, significant alterations in gene expression were observed in the heart, as shown in Table 4 and 5. Among the induced genes, inflammation-related genes were conspicuous.
For example, chemokines are a family of chemotactic cytokines that regulate migration and tissue localization of cells that initiate inflammatory responses. The cytokines participate in inflammatory leukocyte recruitment, in lymphocyte recirculation, and even in cancer metastasis. CXC, one subfamily of the chemokines, has two conserved cysteines separated by one amino acid in the N-terminal residue. In the CXC subfamily, more than 17 ligands have been identified.15 The expression of the CXCL10 [chemokine (CXC motif) ligand 10] gene was dramatically higher in both the hearts and livers of the APAP-treated group (19.2-fold in heart, 21.6-fold in liver) than in the control group. CXCL9 was also significantly induced in the heart (12.9-fold).
IRFs, implicated as regulators of the IFN gene, represent a family with a total of nine members with multiple functions.16,17 IFNs are a family of multi-functional cytokines that mediate cellular resistance against viral infection and also have diverse functions in immune responses to pathogens, immunomodulation, and hematopoietic development. Expression of IFNs is regulated by several IRFs. Following treatment with APAP, IRF-7 was induced 10.1-fold in the heart. IRF-1 and IRF-9 were also induced in the heart (3.1- and 3.0-fold, respectively). IRF-1 has been assigned a dual role in apoptosis, as it serves both pro- and anti- apoptotic functions. In general, IRF-1 activity does not lead to cell death. However, under certain physiological and pathological conditions, it promotes apoptosis. The induction of Casp1 by IRF-1 has been shown to be an essential component of apoptosis induction.16 Transcription of other members of the family of caspases is also induced by IRF-1. Furthermore, IRF-1 can act as a mediator of cytokine-induced apoptosis, and IRF-1 induction appears to be an important signaling event in the initiation of apoptosis by IFN-γ.16,17 Up-regulation of IRF-1 suggests the possibility that apoptotic processes might be induced in the rat heart after APAP intoxication.
Vascular cell adhesion molecule 1 (VCAM-1) is one of the endothelial cell adhesion molecules that mediates leukocyte binding and regulates leukocyte migration from the blood into tissues.18 VCAM-1 activates reduced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase in endothelial cells for generation of ROS, while these ROS activate matrix metalloproteinases (MMPs). Hydrogen peroxide from the VCAM-1 signaling pathway also modulates actin restructuring, causing cytoskeletal changes in endothelial cells. Degradation of the extracellular matrix by MMPs and changes to endothelial cell actin structure are required for VCAM-1 dependent lymphocyte migration. Previous studies have reported that VCAM-1 expression was induced in endothelial cells under inflammatory conditions.18,19 In addition, VCAM-1 was increased in the liver by APAP, reflecting the vascular damage it produced.20 In the present study, the heart VCAM-1 gene was induced 3.8-fold.
Increasing interest is now being shown in the roles of intercellular adhesion molecule-1 (ICAM-1), which functions in the pathogenesis of cardiovascular disease.21 ICAM-1 promotes adhesion between leucocytes and endothelial cells, allowing for tissue-specific localization of leucocytes for immune and inflammatory responses. Cell-cell recognition, leukocyte rolling, and trafficking processes are dependent on cell surface ICAM-1 expression.22 ICAM-1 mRNA expression levels are increased in response to some stimuli, and several inflammatory cytokines and hydrogen peroxide can stimulate ICAM-1 expression.23 ICAM-1 is also increased in the rat myocardium in various models of cardiac ischemia reperfusion injury. The oxidative stress and cytokine release that occur in the infarcted heart are considered to activate NF-κB, while NF-κB activation triggers gene expression of adhesion molecules including VCAM-1 and ICAM-1.24 In the present study, ICAM-1 was induced by APAP in the heart (2.2-fold), but its expression was not altered significantly in the liver. The significance of the induction of inflammation-related genes as a consequence of APAP treatment is very difficult to interpret because the functions of CXCLs and IRFs are varied and intricate.
The induced expression of VCAM-1 and ICAM-1 in heart tissue reflects molecular levels of inflammation and oxidative damage caused by APAP, which were not identified by conventional histopathological measurements. Some cellular stresses, including oxidative stress, were generated in the heart by APAP administration, and consequently, the inflammation-related genes were induced as a means of maintaining homeostasis. Confirmation of this was evident in the significant induction of the Hmox1 gene (6.1-fold), which is a well-known biomarker of oxidative stress in cells. Previous in vivo studies showed that oxidative stress in the liver, represented by an increased Hmox1, was induced by a high dose of APAP.12,13 The up-regulation of Hmox1 by stress-causing agents could mediate cytoprotection against subsequent noxious stimuli, which may be an important physiological process.12 Thus, physiological induction of Hmox1 may represent a protective response to oxidative stress induced by high doses of APAP. The xanthine dehydrogenase gene, which is induced in cells during oxidative stress, was also induced by APAP (2.8-fold).
Based on the induction of these stress genes, oxidative stress was evidently generated in the hearts of rats treated with APAP. In general, oxidative stress may trigger inflammation and ultimately lead to cellular toxicity through the apoptotic process. The imbalance between ROS production and antioxidant mechanisms leads to myocyte dysfunction, injury, and necrosis. ROS can cause lipid peroxidation, resulting in disruption of membrane architecture and subsequent lysosomal enzyme release, while DNA and amino acid oxidation causes genetic mutations and enzyme dysfunctions or proteolysis. Oxidative stress also induces heart remodeling.25
Activated caspases cleave a variety of substrates, including proteins involved in signal transduction (apoptosis regulators, cytokines, serine/threonine kinases, etc.), structural proteins, and proteins involved in the regulation of transcription, translation, and RNA editing.26 The caspases are divided into two subgroups: pro-apoptotic and pro-inflammatory, depending on their major functions. Members of the first group, Casp1-3 and 6-10, have a role in the process of apoptosis and are essential for the activation and implementation of cellular demise.27 Members of the second group, Casp1, 4, 5 and 12, are involved in cytokine maturation and, consequently, the inflammatory response. Active Casp1 is essential for cleavage of pro-interleukin 1β (pro-IL-1β) and pro-IL-18 into their active forms. IL-1β is related to many immune reactions, including the recruitment of inflammatory cells to the site of infection, while IL-18 is important for the production of IFN-γ and enhancement of the cytolytic activity of natural killer cells.28 However, gathering evidence indicates that caspases play roles in many other cellular processes that cannot be classified as pro-apoptotic or pro-inflammatory. The activation of pro-inflammatory caspases can clearly induce apoptosis.26 Caspases are known to play a key role in APAP-induced apoptosis and necrosis of the liver.20 In the present study, among the 11 caspases investigated, Casp1, 4, and 12 were induced in the heart by APAP administration (2.8-, 2.1- and 2.3-fold respectively). Casp4 and 12 were also induced in the liver by APAP (4.4- and 19-fold respectively).
Among 23 fibroblast growth factors (FGFs), only FGF7 was increased (by 2.2 fold) in the heart of the APAP-treated group, compared to the control, but unchanged in the liver. FGF21 and FGF23 were increased in the liver, but not in the heart. The FGFs constitute a family of closely related polypeptides, initially considered to be mitogens. However, several previous studies have reported nonmitogenic actions of FGFs. FGFs can participate as endogenous cardioprotective agents and improve cardiac resistance to ischemia-reperfusion injury.29 FGFs increase cytosolic cGMP levels in a myocardial ischemia-reperfusion setting.30 In turn, cGMP inhibits L-type calcium entry into the cardiomyocytes, preventing calcium accumulation in the mitochondrial matrix and cytosol within the cardiomyocytes. Inhibition of intracellular calcium may shorten phase 2 of the cardiac action potential, providing a negative inotropic effect and decreasing myocardial oxygen and energy consumption. Up-regulation of FGF7 may be a protective response to oxidative stress induced by APAP in the heart, and cGMP and nitric oxide would be involved in this cytoprotective response. In our study, the Nrf2 gene, which is one of the important transcription factors for oxidative stress, was not altered by APAP. Although Nrf2 has been known to be one of the important transcription factors for oxidative stress or detoxification, it is known that Nrf2 induction is not always observed concomitantly with the induction of Nrf2-mediated oxidative stress related genes. Furthermore, decreased expression of Nrf2 gene was reported in the presence of APAP.31
In contrast to the large numbers of the genes induced by APAP in the heart, the number of repressed heart genes was relatively small and most of these genes have yet to be identified. Among those with established identities were many cytochrome p450 genes, including CYP3A, which is known to be actively involved in the metabolism of APAP. The expression ratio was 0.32 in this study. Other groups of cytochrome p450s, such as CYP4A and CYP2J, were also repressed, with ratios of 0.35 and 0.46, respectively. During metabolism of APAP, toxic metabolites including NAPQI were generated; these metabolites may attack the mRNA responsible for translation of metabolic enzymes. Furthermore, toxic metabolites generated by the liver may cause adverse effects when they reach the heart. There are also other potential roles of liver injury and systemic inflammation that can have undesirable effects on heart function. This was not clarified in this study.
In summary, liver injury was induced in rats sacrificed 24 hours after oral administration of 1,000 mg/kg APAP. In contrast, heart intoxication was not identified at the same dosage level by conventional histopathology. However, the induction of many genes related to the processes of oxidative stress, inflammation, and apoptosis was identified in the heart using microarray analysis. These results suggest that APAP may cause unfavorable pathological changes in the heart, and that further studies are needed to identify the molecular mechanisms of cardiotoxicity induced by this frequently used over-the-counter drug.


 XML Download
XML Download