

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Immunoglobulin subtypes are significantly different in their underlying biologic regulation, distribution, and in their interaction with receptors on the various effector cells of the immune system. The immunoglobulin subtypes that are produced in response to a particular immune challenge depend on class-switch gene rearrangement of the constant regions.1 The immunoglobulin G (IgG) class is complex in structure and biology with four subclasses of IgG antibodies, ranging from 1 through 4 based upon the order of their discovery and serum levels. The serum concentration of IgG1 is the highest amongst the IgG subclasses and ranges from 5-11 mg/mL,2 whereas the least abundant subclass, IgG4, is present at mean concentrations of 0.35-0.51 mg/mL.3 The concentration of IgG4 in serum varies significantly among healthy people unlike the other IgG subclasses. IgG4 levels generally range from less than 10 µg/mL to 1.4 mg/mL, with levels over 2 mg/mL noted in rare instances and levels being generally higher in men and older people.2-4
IgG4 is a unique antibody based upon its structure, function, and immunologic regulation. IgG4 antibodies are distinct as they undergo 'half-antibody exchange' in vivo, resulting in recombined antibodies comprising two different binding specificities.5 IgG4 does not activate complement pathways and has reduced effector function relative to other IgG subtypes.6,7 IgG4 production is driven in part by T-helper cell 2 cytokines.8,9 IgG4 plays a significant role in bullous skin lesions,10 atopic eczema and bronchial asthma11,12 as well as decreasing IgE-mediated inflammatory response in parasitic infections.8,9 IgG4-related systemic disease (IgG4-RSD) represents a newly described category of diseases with a potential role for IgG4 antibodies. The exact role of IgG4 antibodies in the pathogenesis of IgG4-RSD still remains unclear in the present day scenario. An increased understanding of the precise role of IgG4 in these related clinical syndromes will be paramount for elucidation of the underlying pathophysiology.
HISTORICAL PERSPECTIVE OF AUTOIMMUNE PANCREATITIS AND IgG4-RELATED DISEASES
Sarles, et al.13 raised the possibility of some cases of chronic pancreatitis arising from an autoimmune pathologic process in 1961. The concept of autoimmune pancreatitis (AIP) was proposed by Yoshida, et al.14 in 1995 along with the introduction of this terminology. Autoimmune pancreatitis has been recognized over the years as a distinct clinical entity, characterized by a mass lesion in the pancreas, painless obstructive jaundice, pancreatic duct narrowing, association with diabetes mellitus and favorable response to steroid therapy.
The autoimmune association of AIP is based upon its association with other immune-mediated diseases, such as sclerosing cholangitis, primary biliary cirrhosis (PBC), Sjogren syndrome, and inflammatory bowel disease (IBD).15,16 Autoantibodies, including antinuclear antibodies, rheumatoid factor, anticarbonic anhydrase II, and antilactoferrin are commonly encountered in patients with AIP.17-20 Kawa, et al.21 reported an association with HLA DRB1*0405-DQB1*0401 haplotype in a study from Japan. Numerous terminologies have been coined by different investigators to emphasize selected aspects of the disease, including chronic sclerosing pancreatitis,22 lymphoplasmacytic sclerosing pancreatitis,23 nonalcoholic duct-destructive chronic pancreatitis,24 idiopathic tumefactive chronic pancreatitis25 and duct-narrowing chronic pancreatitis.26 Over the past decade, developments have resulted in the establishment of the theory that AIP is a heterogeneous entity which includes at least 2 distinct clinicopathologic entities: 1) lymphoplasmacytic sclerosing pancreatitis (LPSP), and 2) idiopathic duct-centric chronic pancreatitis (IDCP).19,27,28
LPSP is a histologically unique lesion that was proposed by Kawaguchi, et al.23 in 1991. It consists of diffuse lymphoplasmacytic infiltration and fibrosis that focally gives rise to a storiform fibrosis. Neutrophils are conspicuously absent but eosinophils may be identified. Pancreatic lobules are relatively well preserved compared to alcoholic chronic pancreatitis, but focal destruction of pancreatic acini and replacement with fibrosis are routinely noted. This inflammatory process also characteristically extends around the main and interlobular ducts, leaving the duct epithelium and lumen intact. Veins are almost always obliterated by the same inflammatory process (obliterative phlebitis). Even the splenic and portal venous vasculature may be involved, leading to an intraoperative differential diagnosis of carcinoma. The common bile duct is usually inflamed resulting in jaundice seen in patients with AIP. Numerous IgG4-positive plasma cells are identified in LPSP on staining.29,30 LPSP predominantly affects elderly men with a mean age of 62 to 63.4 years, is commonly associated with proximal biliary tract, retroperitoneal, renal, and salivary disease, shows elevated serum IgG4 titer and relapse (47%) in a significant proportion of cases.31
IDCP is characterized by ductal epithelium centric inflammation.16,27,32 Neutrophilic infiltration in the main and/or interlobular ducts is typically seen, and extends to involve the duct epithelium and lumen. The duct epithelium shows destructive and regenerative changes with the lumen appearing stenotic or tortuous as a result of the inflammation. A band of lymphocytes and plasma cells surrounds the lumen but, in contrast to LPSP, the ductal lesion lacks the appearance of a thickened wall. The entire duct may seem entrapped within an aggregate of inflammatory cells. When the inflammation is severe, pancreatic lobules are also inflamed with neutrophils, lymphocytes and plasma cells and microabscesses may be seen as well. Although there is fibrosis around pancreatic lobules, inflammatory cells are scarce within fibrosis itself, in contrast to LPSP, in which inflammatory cells are numerous within fibrosis. Obliterative phlebitis is rare, and inflammation of the common bile duct is less common compared to LPSP. IgG4-positive plasma cells are usually few in IDCP.32
The clinical features of LPSP are concordant with those of AIP reported from Japan, described previously.27 Serum IgG4 is elevated in 80% of AIP patients in Japan, which correlates well with numerous IgG4-positive plasma cells seen in LPSP. In contrast, patients with IDCP are younger than LPSP patients, and many of them are younger than 40 years without any specific gender preponderance.27 Obstructive jaundice is less common in IDCP than in LPSP. The association of IBD is found in IDCP, but extrapancreatic manifestations seen in LPSP are rare. It is noteworthy that IDCP is rare in Japan.33
Owing to the fact that both LPSP and IDCP have overlapping clinicopathological features, there has been a contentious debate as to whether these two pathological groups are different manifestations of a single entity of AIP, or whether they are distinct clinicopathological entities. The controversy is due to the variety of AIP diagnostic criteria proposed by different groups. The diagnostic criteria from Japan,34 Korea,35 consensus of the Japan-Korea symposium36 and Mayo Clinic37 define LPSP as the pathological entity of AIP, but other groups include both LPSP and IDCP in AIP.16,32,38 The theory that LPSP and IDCP are distinct entities has gained acceptance due to the differing demographic and clinical presentations as well as different immunoreactivity for IgG4. Recently, new nomenclature, type 1 and type 2 AIP, which correspond to LPSP and IDCP, respectively, have been proposed.39
IgG4-RELATED SCLEROSING PANCREATITIS
In 2001, Hamano, et al.40 reported elevated serum IgG4 levels in a cohort of patients with a specific subtype of sclerosing pancreatitis/AIP. This finding was not observed in patients with pancreatic carcinoma, primary sclerosing cholangitis (PSC), PBC, nonspecific chronic pancreatitis, Sjogren syndrome and normal subjects. Kamisawa, et al.41 reported that IgG4-positive plasma cells are increased systemically in patients with AIP and concluded that AIP patients have a systemic disease, with the proposal for the entity 'IgG4-related sclerosing disease'.42 Histologically, this subtype corresponds to LPSP, but not IDCP.30,32,43
CLINICAL FEATURES OF IgG4-RELATED SCLEROSING PANCREATITIS
IgG4-related sclerosing pancreatitis is seen most commonly in middle-aged and elderly men with a mean age of 59 to 68 years and a male-to-female ratio of 4-7.5: 1.44-46 The prevalence rate is 2% to 11% among patients with chronic pancreatitis.20,45 Studies have demonstrated that it accounts for up to one third of the total cases of benign conditions that are treated with pancreatoduodenectomies, thus highlighting the fact that is often mistaken for pancreatic cancer in spite of major advancements in clinical medicine and radiology.47,48
Patients usually present with a pancreatic mass and/or painless obstructive jaundice. Newly diagnosed type II diabetes mellitus and steatorrhea are noted in some of these cases.20,37 Contrarily, systemic symptoms, including fever, weight loss, and generalized malaise are rarely seen. Imaging studies characteristically show enlargement of the pancreas, predominantly involving the pancreatic head, and endoscopic retrograde cholangiopancreatography (ERCP) shows irregular narrowing of the pancreatic duct with or without stenosis of the common bile duct.36
The disease responds well to steroid therapy, although relapses can occur on cessation of treatment.37 Response to treatment is monitored by a decline in serum IgG4 titer and a reduction in IgG4+ cells in the affected organs. Rituximab administration may be significantly beneficial according to a recent study which reports an excellent response in IgG4-related pancreatitis cases.49 The drug acts by reducing the number of B-lymphocytes that provide a source for the IgG4-secreting plasma cells.
MORPHOLOGIC FEATURES OF IgG4-RELATED SCLEROSING PANCREATITIS
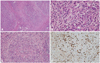
The cut surface of the pancreas is firm with tan-white areas interspersed amidst the lobulated yellow-gray parenchyma (Fig. 1). The histologic triad necessary for a diagnosis comprises of the following: 1) lymphoplasmacytic infiltrate, 2) sclerosis, and 3) obliterative phlebitis, with accompanying atrophy and destruction of pancreatic acini although these morphologic features are characteristic, they are not completely specific, and thus a definitive diagnosis requires the demonstration of an increased population of IgG4+ plasma cells.31
Sclerosis of the interlobular septa further enhances the lobular architecture of the pancreas by making the separate lobules appear more demarcated (Fig. 2A). The extent of involvement varies greatly from one lobule to another. This may range from normal appearing lobules, to moderate atrophy and inflammation, and complete replacement by a fibroinflammatory process. The pancreatic lobules show interstitial infiltration by plasma cells, reactive lymphocytes, and a variable number of eosinophils, accompanied by atrophy or loss of acini (Fig. 2B). The lobular stroma shows variable degrees of sclerosis, which can exhibit a storiform character.36,37 Prominent edema or myxoid change may be noted. Increasing degrees of sclerosis results in loss of the lobular architecture.50 The fibrosclerosis and inflammatory process can extend beyond the confines of the pancreas into the retroperitoneal tissue and surrounding organs. Islet cell hyperplasia is a rare finding in contradistinction to the routinely encountered examples of chronic pancreatitis. The pancreatic ducts appear more prominent as a result of being surrounded by a periductal cuff of chronic inflammatory cells, accompanied by a sclerosing stromal reaction. The ductal epithelium is not infiltrated by inflammatory cells. Irregular narrowing of the ductal lumen gives rise to the characteristic imaging findings.
Phlebitis, characterized by transmural inflammatory infiltration of the veins, with or without fibrous obliteration of the lumen, is a sine qua non of this disease (Fig. 2C).31 An elastic stain may highlight the venous obliteration. Lymphoid follicles can be present in the stroma. Sometimes fibroinflammatory mass lesions are formed, which have been called "inflammatory pseudotumor" in the literature.51 Another noteworthy feature often not reported is the tendency for the inflammatory cells to form cuffs around nerves. Plasma cells and lymphocytes can even infiltrate into the nerves.31
DIAGNOSIS OF IgG4-RELATED SCLEROSING PANCREATITIS AND IgG4-RELATED SCLEROSING DISEASE
A preoperative diagnosis of IgG4-related sclerosing pancreatitis is necessitated by the need to avoid surgery in these cases which are otherwise treated with steroids.52 Although there is no definite cut-off limit for the number of IgG4+ plasma cells currently, greater than 30 IgG4+ plasma cells per high-power field has been reported as being specific.30,32 A recent study suggests a cut-off level of more than 50 per high-power field IgG4-positive cells to be more specific (Fig. 2D).53 In nonspecific chronic inflammation or pancreatic carcinoma, few scattered IgG4+ cells are seen. The number of IgG4+ plasma cells is more sensitive and specific than serum IgG4 titer as the latter is not elevated in approximately one third of cases but may be elevated in other conditions such as in patients with pancreatic carcinoma.54-56 Alternatively, the ratio of IgG4+/IgG+ cells (>40%) can be used as a standard parameter to arrive at a sensitive and specific diagnosis.43,57,58
The diagnosis of IgG4-related sclerosing pancreatitis is usually rendered upon consideration of clinical, laboratory, imaging, and histologic features. Biopsies may be falsely negative since the pancreas may not be involved in its entirety.59 ERCP guided biopsies of the pancreas may be more helpful in demonstrating the presence of IgG4+ plasma cells.60 The diagnostic criteria have undergone several changes and vary from one continent to another (Table 1 and 2). However, the diagnosis is almost always determined by the discovery of increased number of IgG4-positive plasma cells coupled with the morphologic findings. The diagnosis can be rendered possible in the absence of biopsy tissue with the aid of imaging studies, extrapancreatic organ involvement, and response to steroids may help in establishing the diagnosis if tissue samples are not available.36,37,61
IgG4-related sclerosing pancreatitis is frequently associated with extrapancreatic lesions, and the spectrum of disease manifestations involving different organs has continued to expand.20,37,61-72 Hamano, et al.62 reported that extrapancreatic bile duct lesions occur in 73.9%, hilar lymphadenopathy occurs in 80.4%, lacrimal and salivary gland lesions in 39.1%, hypothyroidism in 22.2%, and retroperitoneal fibrosis in 12.5% of cases with IgG4-RSD. The various sites of involvement exhibit similar morphologic features with the common denominator being a significant increase in the number of IgG4+ plasma cells (Table 3).29,41,42
Extrapancreatic involvement may develop prior to, concomitantly or following pancreatic disease, or may even occur independently in the absence of pancreatitis. A so called association of "autoimmune pancreatitis" with other "autoimmune diseases" including PSC, Sjogren syndrome, and retroperitoneal fibrosis simply represents extrapancreatic manifestations of a single systemic entity rather than a multitude of diseases.39,73,74 Another pointer favoring this theory states that an increased number of IgG4+ plasma cells can be identified in other uninvolved organs in such cases organs, such as the aerodigestive tract mucosa and bone marrow, thus providing credence in favor of the systemic nature of this disease.42,65,75,76 The various names ascribed to this disease include IgG4-related sclerosing disease,77 IgG4-related systemic sclerosing disease,78 IgG4-related disease,61,79 IgG4-related autoimmune disease,42 hyper-IgG4 disease,80 and IgG4-related systemic disease.49
The extrapancreatic lesions of IgG4-related sclerosing disease also exhibit the same characteristic histologic features including lymphoplasmacytic infiltrate, sclerosis, and obliterative phlebitis as similarly seen in IgG4-related sclerosing pancreatitis. The relative proportion of the lymphoplasmacytic infiltrate and sclerosis in different cases gives rise to a spectrum of histologic patterns: pseudolymphomatous, mixed, and sclerosing. These patterns represent different stages in the evolution of this disease with the sequence progressing from being initially pseudolymphomatous to mixed and subsequent progression to the sclerosing pattern. The pseudolymphomatous pattern is more commonly identified in superficial locations including lacrimal gland, salivary gland, breast, and skin and thus these are detected earlier than deep seated sclerotic lesions in locations such as the retroperitoneum.31
The pseudolymphomatous pattern is characterized by a mass forming dense infiltrate of small lymphocytes and plasma cells, with interspersed reactive lymphoid follicles and a variable proportion of scattered eosinophils. The differential diagnosis includes low grade non-Hodgkin's lymphoma, particularly extranodal marginal zone lymphoma. However, these can be distinguished based on the absence of lymphoepithelial lesions, monocytoid B-cells, diffuse sheets of CD20+ B cells, light chain restriction, aberrant immunophenotype, and clonal immunoglobulin gene rearrangements.31
The mixed pattern is the most common, comprising of patchy dense infiltrates of small lymphocytes and plasma cells that are accompanied by significant sclerosis (Fig. 2B). Variable numbers of eosinophils and reactive lymphoid follicles are usually seen. The sclerosing pattern is characterized by a predominantly sclerotic process with poorly defined borders and irregular extensions into the surrounding tissues. Patchy aggregates of lymphocytes and plasma cells, with or without lymphoid follicle formation, are seen diffusely scattered over the extent of the lesion, and are seen more commonly at the periphery than at the center of the lesion. The sclerotic areas comprise of collagen deposition with scattered fibroblasts. Obliterative phlebitis is often seen except in small biopsies.31 The main differential diagnoses are nonspecific inflammation with fibrosis, and malignancies with sclerosis including nodular sclerosing Hodgkin lymphoma, sclerosing large B-cell lymphoma, primary or metastatic carcinoma with desmoplastic stromal changes. The veins show segmental or circumferential transmural infiltration by chronic inflammatory cells, and can show luminal occlusion by fibrous tissue.31 Obliterated veins can be difficult to recognize and may necessitate the aid of elastin stains for identification purposes.
Hepatobiliary system
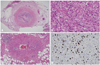
Biliary tract involvement (IgG4-related sclerosing cholangitis) is seen in 50% to 90% of patients with IgG4-related sclerosing pancreatitis and clinically presents as obstructive jaundice or fever.23,51,81-84 Biliary tract disease may also be seen in some cases without pancreatic involvement. Both the extrahepatic and intrahepatic bile duct systems can be affected with the prior being predominantly involved in most reported cases.85 As seen with AIP, the differential diagnosis includes a malignancy of the bile duct system.81-85 The involved bile ducts demonstrate a transmural lymphoplasmacytic infiltrate with variable numbers of eosinophils, sclerosis (Fig. 3A), and obliterative phlebitis (Fig. 3B). The bile duct lumina undergo stenosis owing to the periductal fibrosis and inflammation (Fig. 3C) and the peribiliary glands are often involved in this region.85 Immunohistochemical staining for IgG4 demonstrates a significant subset as being IgG4-positive plasma cells (Fig. 3D).
The clinical and pathologic features overlap with those seen in PSC, a chronic biliary inflammatory disease of unknown etiology. The pathologic process in PSC also involves prominent ductal and periductal fibrosis, luminal stenosis, obliteration, and dilatation.86 It is thought that some of the cases reported as PSC actually represent IgG4-related sclerosing cholangitis.87-91 Mendes, et al.92 published a series of cases of PSC wherein elevated serum IgG4 was found in 9% of the patients and this group of patients also had a lower frequency of IBD.
In IgG4-RSD involving the bile ducts, the bile duct epithelium is usually spared but the peribiliary glands exhibit a greater degree of involvement and inflammation tends to be more severe toward the adventitial aspect. Onion-skin type concentric periductal fibrosis, which is a diagnostic hallmark of PSC, is not seen in IgG4-related cholangitis.81-84 Another distinguishing feature is that a large number of IgG4-positive plasma cells are identified in IgG4-related cholangitis but not in PSC. PSC is a progressive disease culminating in cirrhosis with liver failure requiring transplantation, whereas IgG4-related sclerosing cholangitis is treated with steroid therapy in most cases.81-84,93
The gall bladder may be involved in up to one fourth of patients with IgG4-related sclerosing pancreatitis with the most common manifestation being acute acalculous cholecystitis.94 Co-existing IgG4-related sclerosing cholangitis is also frequently encountered. IgG4-related cholecystitis shows predominant extramural involvement with adventitial inflammation and subserosal inflammatory nodules in contrast to the nonspecific acalculous cholecystitis, which is seen occasionally in patients with PSC, pancreatic cancer, and biliary calculi.58,95
Hepatic involvement is also frequently identified with IgG4-related cholangitis or pancreatitis. The commonest presentation is seen as a mass lesion often referred to as an "inflammatory pseudotumor".51,96,97 The lesion usually involves the hilum of the liver and is bile duct centric with the usual diagnostic triad of a lymphoplasmacytic infiltrate, sclerosis, and obliterative phlebitis. IgG4-related hepatic "inflammatory pseudotumor" might represent an extension of IgG4-related sclerosing cholangitis into surrounding hepatic tissue.31,51,96,97
This IgG4-related lesion is morphologically different from soft tissue related lesions including inflammatory pseudotumor/inflammatory myofibroblastic tumor of the liver since it does not comprise of a spindle cell or myofibroblastic proliferation, does not express the ALK protein frequently associated with inflammatory myofibroblastic tumor and instead comprises of numerous IgG4+ plasma cells with surrounding sclerosis and phlebitis.98 Follicular dendritic cell sarcoma is another entity which must be included in the differential diagnosis of these lesions but can be distinguished by the presence of intersecting fascicles of spindle cells as well as positivity for follicular dendritic cell markers (CD21 and CD35), and Epstein-Barr virus in situ hybridization studies.31,99
Different morphologic patterns are appreciated in liver biopsies obtained from patients with IgG4-RSD. These include portal inflammation with or without interface hepatitis, portal sclerosis, large bile-duct obstructive features, lobular hepatitis, and canalicular cholestasis.100 The number of IgG4+ plasma cells is concomitantly increased and helps to distinguish IgG4-related hepatic disease from other differentials including PSC, autoimmune hepatitis, primary biliary sclerosis, and chronic viral hepatitis in which only far fewer IgG4+ plasma cells are identified.100,101
Salivary gland
The submandibular gland is the salivary gland most frequently involved by IgG4-RSD and is also referred to as 'chronic sclerosing sialadenitis' or 'Kuttner tumor'. The disease was originally described by H. Kuttner102 in 1896, who reported it as a unilateral or bilateral "hard swelling" of the submandibular glands. Chronic sclerosing sialadenitis demonstrates a predilection for the submandibular gland, although parotid gland involvement has also been reported.103 It is characterized by the presence of a prominent lymphoplasmacytic infiltrate and lymphoid follicles, which upon being combined with the finding of cytotoxic T cell populations, indicate an immunological process.104
Clinical manifestations include unilateral or bilateral hard masses that are often mistaken for a tumor based on both clinical and radiological features.57,61,64,105-107 Almost all cases of chronic sclerosing sialadenitis have been shown to be IgG4-related in the submandibular gland, whereas up to a third of all cases of nonspecific chronic sialadenitis are IgG4-related.57,61,64 Recognition of IgG4-RSD in the salivary glands is significantly important as these patients may develop IgG4-related lesions in other body sites as well. The parotid glands can be involved either solely or along with the submandibular glands. The morphologic features of salivary gland involvement are identical to those observed in other organs.57,61,64 It is characterized by sclerosis of interlobular septae with resulting accentuation of the salivary gland lobular architecture and an interstitial infiltrate of lymphocytes and plasma cells along with variable degrees of sclerosis. The involvement of adjacent salivary lobules may be non-uniform with focal atrophy and loss of acini in some lobules. Reactive lymphoid follicles and collagenous sheaths around small ducts are frequently seen.57,61,64
IgG4-related sclerosing dacryoadenitis and/or sialadenitis is one of the major causes of Mikulicz disease with bilateral lacrimal and salivary glands involvement.108-110 Mickulicz disease, which can represent a number of clinical etiologic entities, is defined clinically as bilateral, painless, and symmetrical swelling of the lacrimal, parotid, and submandibular glands of unknown etiology for a duration exceeding 3 months. It is not synonymous with the entity Sjogren syndrome, a distinct autoimmune disease featuring keratoconjunctivitis sicca, xerostomia, intermittent swelling of the salivary or lacrimal glands, presence of anti-Ro/SSA and anti-La/SSB autoantibodies, and morphologically characterized by lymphoepithelial sialadenitis.110
Orbit, including lacrimal glands
IgG4-RSD commonly affects the lacrimal glands and surrounding orbital soft tissues and these were the initial sites reported to be involved by this disease apart from the pancreas. The patients usually present with unilateral or bilateral, painless orbital swellings of long duration with or without significant impairment of visual acuity or symptoms of 'dry eye'.60,65 The differential diagnosis on clinical and radiological examination includes inflammatory pseudotumor and lymphoma. Extensive involvement of orbital soft tissues and sclerosis may lead to markedly decreased visual acuity and occasionally cause optic nerve atrophy and blindness.65 Steroid therapy is the primary treatment but may fail to ameliorate the condition in severe cases with irreversible sclerosis and fibrosis of orbital tissues.60,65
The morphologic features of lacrimal gland involvement (IgG4-related chronic sclerosing dacryoadenitis) are similar to those of IgG4-related sclerosing disease in other organs, except that obliterative phlebitis is not as commonly seen.60,65 Progressive sclerosis and gland destruction frequently comprise the course of this disease in the orbit with an eventual burnout phase where only the fibrosis remains in the absence of increased inflammation. It may cause extensive fibrosis to the degree that no recognizable lacrimal gland or orbital structure is identified on biopsies. All patterns of inflammation and fibrosis, including pseudolymphomatous, mixed and sclerotic patterns, are identified in the lacrimal gland and orbital tissues.31,60,65
Retroperitoneum and mesentery, mediastinum, and aorta
Inflammatory fibrosclerosing lesion involving the retroperitoneum, mesentery and mediastinum is an idiopathic entity characterized by diffuse fibrosis and is confused with inflammatory pseudotumor or inflammatory myofibroblastic tumor.111 Idiopathic retroperitoneal fibrosis or mediastinal fibrosis may be associated with Riedel thyroiditis, orbital/lacrimal IgG4-RSD or IgG4-related sclerosing cholangitis.112 The fibrosis in these locations may extend to involve the kidney, ureter and bowel mesentery with varying response to steroid therapy which is ineffective in advanced fibrosis.113-118
Noninfectious aortitis is represented by a group of inflammatory disorders characterized by chronic inflammation within the aortic wall, wherein the evident inflammation is not considered to be due to infection.119 Noninfectious aortitis is most commonly seen involving the thoracic aorta, especially the ascending aorta, and may result from well known systemic rheumatologic disorders such as giant cell arteritis, Takayasu arteritis, rheumatoid arthritis, ankylosing spondylitis, and Behcet's disease.120,121
The lesions described above are often characterized by a variably prominent lymphoplasmacytic infiltrate, sclerosis, and phlebitis which are essentially identical to those of IgG4-RSD.122 A frequent association with pancreatitis has also been reported in several cases.118,123 Upon extrapolating these findings, a significant proportion of cases of retroperitoneal fibrosis,124,125 mediastinal fibrosis,67 sclerosing mesenteritis,126 periaortitis and inflammatory aortic aneurysm (with cases predominantly involving abdominal aorta with rare cases involving aortic arch)71,127-132 have been demonstrated to express markedly increased numbers of IgG4+ plasma cells, and hence represent members of IgG4-RSD. It has not been determined at present whether the group with no significant increase in IgG4+ cells has different etiologies or represents burnt out IgG4-RSD.31
Thyroid
Involvement of the thyroid gland as part of the spectrum of IgG4-RSD is being reported increasingly. Patients with AIP often have concomitant evidence of hypothyroidism, with increased levels of antithyroglobulin antibody.133 The exact pathogenetic mechanism for this phenomenon has yet to be elucidated. A study by Li, et al.134,135 involved segregating 70 cases of Hashimoto thyroiditis into 2 groups: 19 IgG4-related cases and 51 non-IgG4-related cases. The IgG4-related group comprised of cases with a greater degree of stromal fibrosis, lymphoplasmacytic infiltration, and thyroid follicular cell destruction than the other group. The IgG4-related subset is associated with a higher level of circulating thyroid autoantibodies, lower female-to-male ratio and an aggressive clinical course. It is possible that hypothyroidism in patients with autoimmune pancreatitis may result from an IgG4-related thyroiditis which may be mistaken for Hashimoto thyroiditis in view of the overlapping symptoms and laboratory findings. Riedel thyroiditis is also regarded as a component of the multifocal fibrosclerosis seen in IgG4-RSD with reports backing this theory based on immunohistochemical findings.136-138
Lung and pleura
Lung involvement by IgG4-RSD may present insidiously with the usual pulmonary symptoms including cough, chest pain, dyspnea and hemoptysis.69,139-141 Radiologically, it may appear as solitary or multiple lung nodules, or consolidation and hilar lymphadenopathy.142 Distinct patterns of pulmonary involvement include: solid nodules, peribronchial or perivascular, and interstitial alveolar infiltrates.140
The solid nodular type comprises a hilar or peripheral mass forming lesion with the characteristic lymphoplasmacytic infiltrate and accompanying sclerosis.69 The adjacent alveoli show interstitial infiltration of lymphocytes and plasma cells. In cases involving the hilum, sclerosing inflammation of the bronchial wall is identified and it is almost always accompanied by involvement of the bronchial glands. The bronchovascular type is characterized by lymphoplasmacytic infiltrate distributed along the lymphatic channels, with interstitial expansion and extension along the bronchovascular bundles, interlobular septa, and pleura. Lymphatic dilatation with histiocytes showing emperipolesis of lymphocytes is a helpful identifying feature.139 In the alveolar interstitial type, the lymphoplasmacytic infiltrate is limited to the alveolar interstitium and is difficult to distinguish from nonspecific interstitial pneumonia.
A variable population of eosinophils along with lymphoid follicles is routinely seen in up to 50% of these cases. One distinguishing feature noted in pulmonary cases of IgG4-RSD is that the arteries are also frequently involved and obliterated as opposed to other body sites wherein veins are selectively involved.139,140 Endothelialitis characterized by a lymphoplasmacytic infiltrate in the subendothelium leading to narrowing and obliteration of the vascular lumen is often identified. Visceral or parietal pleural involvement is noted usually in the form of fibrous thickening accompanied by a chronic lymphoplasmacytic infiltrate with or without fibrinous exudates.139-141
Breast
IgG4-related sclerosing mastitis can present as single or multiple unilateral or bilateral painless masses, with or without evidence of systemic IgG4-RSD.70,143 Morphologically, it is characterized by a prominent lymphoplasmacytic infiltrates with lymphoid follicle formation, patchy fibrosis, and atrophy of breast lobules. Phlebitis is also seen in some cases. The differential diagnosis includes involvement of the breast by lymphoma owing to the dense lymphoplasmacytic infiltrate or hyaline-vascular variant of Castleman disease if follicles with regressive changes are seen.31
Kidney and urinary tract
IgG4-RSD may also involve the kidneys and urinary tract leading to disturbances in renal function.144-147 Radiologic studies may demonstrate the presence of poorly circumscribed nodular masses in the kidneys, often necessitating the exclusion of malignancy.145 The commonest findings on biopsy or nephrectomy are tubulointerstitial nephritis with an IgG4-positive plasma cell-rich inflammatory infiltrate, fibrosis, and tubular atrophy. IgG4 electron-dense deposits have been demonstrated in the peritubular basement membrane using electron microscopy. The renal glomeruli are usually uninvolved but few cases with concurrent membranous nephropathy have been recorded.144-148 The tubulointerstitial nephritis is steroid responsive, whereas the response of membranous glomerulonephritis can be variable. IgG4-related chronic sclerosing pyelitis and IgG4-associated inflammatory pseudotumor of the ureter presenting as hydronephrosis have also been reported (Fig. 4).149,150
Central nervous system
Central nervous system involvement by IgG4-RSD is extremely rare, and the most commonly reported site is the pituitary gland (IgG4-related infundibulo-hypophysitis). A male predominance is noted. The clinical picture includes patients presenting with hypopituitarism, diabetes insipidus, and/or symptomatic local mass effect.151-156
A pituitary mass and/or thickening of the pituitary stalk are identified on imaging studies. The treatment modality is steroid therapy which usually elicits a favorable response and manifests as a marked reduction in lesional size and correction of hormonal imbalances.151-156 An accompanying meningeal involvement in the form of hypertrophic pachymeningitis or sinus involvement, with extension to the sellar and parasellar regions is identified in some cases.152 The pituitary shows patchy or extensive interstitial infiltration of lymphocytes and plasma cells, along with loss of pituitary parenchyma.
Rare cases of dural spinal involvement have been reported (IgG4-related sclerosing pachymeningitis).157 Such lesions described as idiopathic hypertrophic pachymeningitis may represent part of the spectrum of IgG4-RSD.158,159 It is still unclear whether a few reported cases of intracranial inflammatory pseudotumor with increased IgG4+ plasma cells reported by Lui, et al.160 actually represent IgG4-RSD since these patients did not exhibit any other manifestations of systemic IgG4-RSD.
Skin and other miscellaneous body sites
Skin involvement in IgG4-RSD may be either isolated or as part of systemic IgG4-RSD.68,161 The clinical presentation is in the form of cutaneous plaques or nodules tending to involve the head and neck. Microscopic examination reveals a dermal and subcutaneous nodular lymphoplasmacytic infiltrate with formation of lymphoid follicles and dermal or subcutaneous sclerosis. A markedly increased number of IgG4+ plasma cells, small lymphocytes, and variable infiltrate of eosinophils is identified.31
Cases of IgG4-RSD have been reported in the pericardium,162 prostate,163 seminal vesicles,164 nose, paranasal sinuses165,166 and uterine cervix (idiopathic cervical fibrosis).167 Questionable entities currently include autoimmune esophagitis and splenic sclerosing angiomatoid nodular transformation, which exhibit some of the features overlapping with IgG4-RSD, including increased plasma cells and sclerosis.168-171 However, there is no definite evidence to currently include these lesions under the spectrum of IgG4-RSD.
Lymph nodes
Lymphadenopathy is identified in up to 80% of patients with autoimmune (IgG4-related sclerosing) pancreatitis as evidenced by imaging studies.62 Lymph node involvement may be multifocal with the commonest nodes involved being mediastinal, abdominal, and axillary lymph nodes. Clinical manifestations vary greatly.66,161,172 Enlarged asymptomatic lymph nodes are identified in surgical resection specimens or identified on imaging studies of patients with known IgG4-RSD. Lymphadenopathy may also develop following an established diagnosis of extranodal IgG4-RSD or may be the initial presenting manifestation with subsequent discovery of systemic IgG4-RSD. The enlarged lymph nodes may be asymptomatic or may give rise to a mass effect in various body sites.
The differential diagnosis in these cases with lymphadenopathy includes lymphoma, multicentric Castleman disease, or metastatic malignant tumor. The lymph nodes are usually not exceedingly enlarged (≤2 cm) and not associated with fever and weight loss. Serum lactate dehydrogenase level is normal or only slightly elevated. Other laboratory findings include raised serum IgG4, serum IgG, polyclonal hypergammaglobulinemia and elevated erythrocyte sedimentation rate (ESR).66,161
The morphologic features of IgG4-related lymphadenopathy are different from those of extranodal sites since there is usually no sclerosis or phlebitis. Five histologic patterns of IgG4-related lymphadenopathy have been described.66,79,161 The various subtypes are as follows: multicentric Castleman disease-like pattern (type I),172-178 follicular hyperplasia pattern (type II),66 interfollicular plasmacytosis and immunoblastosis pattern (type III),66,79,161 progressive transformation of germinal center-like pattern (type IV),79,179 and nodal inflammatory pseudotumor-like pattern (type V).79,179,180 Abundant IgG4+ polyclonal plasma cells are present in interfollicular and follicle center compartments in all of these patterns (Fig. 5).79
IgG4-RELATED SCLEROSING DISEASE AND ASSOCIATED MALIGNANCIES
Sato, et al.79 have reported cases of ocular marginal zone lymphoma associated with ocular adnexal IgG4-related sclerosing disease. A few cases of localized non-Hodgkin's lymphoma (marginal zone lymphoma and follicular lymphoma) arising in a background of IgG4-related chronic sclerosing dacryoadenitis have been recently reported.181 A rare example of composite marginal zone lymphoma and classical Hodgkin lymphoma associated with IgG4-related sclerosing disease of the cervical soft tissue has also been published.167 A recent study from the Mayo Clinic cites 3 cases of lymphoma (2 large B-cell lymphomas and 1 B-cell lymphoma unspecified) discovered on follow-up of 111 patients with IgG4-related sclerosing disease, suggesting that the syndrome increases the risk of developing lymphoma in various body sites.182
Several cases of pancreatic ductal adenocarcinoma have been published in association with IgG4-RSD.183-186 Case reports of pulmonary adenocarcinoma, salivary duct carcinoma, urothelial carcinoma in situ and gastrointestinal clear cell sarcoma in association with IgG4-RSD have been reported in the lung, parotid gland, ureter and small intestine, respectively.140,150,187,188 Further studies are mandated in this field to determine if there is any causal relationship between the malignancies and IgG4-RSD.
PROPOSED ETIOPATHOGENESIS OF IgG4-RELATED SCLEROSING DISEASE
The pathogenesis of IgG4-related sclerosing disease is still not clearly understood. The disease may represent a hypersensitive/allergic reaction as compared to being an autoimmune disease.189 An association of IgG4-related AIP with gastric ulcer and Helicobacter pylori (H. pylori) infection has been proposed.190-192 It has been theorized by Okazaki, et al.193 that the development of the acronym 'AIP' involves a biphasic mechanism in which the initial response to self-antigens such as carbonic anhydrase, lactoferrin, pancreatic secretory trypsin inhibitor and ά-fodrin and molecular mimicry (H. pylori) are induced by decreased naive regulatory T cells, in conjunction with T-helper 1 cells releasing proinflammatory cytokines. Progression is mediated through increased memory regulatory T cell and T-helper 2 cell immune responses resulting in chronic inflammation. The inflammatory infiltrate usually comprises of a mixed population of T and B cells, with increased CD4+, CD25+, FOXP3+ regulatory T cells.31,194,195 T-helper cell 2 cytokines (interleukin-4, interleukin-5, and interleukin-13) and regulatory cytokines (interleukin-10 and transforming growth factor) are upregulated in the involved tissues.194-196 Other proposed hypothesis regarding the pathogenesis include enhanced T-helper type 2 responses to intestinal microflora197 and an immune-mediated pathogenesis based on the ultrastructural finding of electron dense immune complex deposits along the basement membranes of pancreatic acini and renal tubules.32
According to the results of a recent study conducted by screening of pooled IgG from patients with AIP, a specific reactivity with a peptide demonstrating homology with plasminogen-binding protein of H. pylori and with ubiquitin-protein ligase E3 component n-recognin 2 (an enzyme with high levels of expression in pancreatic acinar cells) was noted in the serum samples from 90% of patients with AIP and 10% of patients with pancreatic cancer. Healthy controls did not show any expression.198 It remains to be seen whether H. pylori truly plays a role in the development of AIP and IgG4-RSD.
DIAGNOSTIC CRITERIA FOR IgG4-RELATED SCLEROSING DISEASE
Diagnosis of IgG4-RSD requires not only an increase in the absolute number of IgG4+ cells but also an increased IgG4+/IgG+ ratio for the correct diagnosis. An absolute IgG4+ plasma cell count is not sufficient as a sole index as IgG4+ cells normally comprise approximately 5% of all IgG+ cells, and may be present in significant numbers in any inflammatory lesions with increased plasma cells even though the percentage might remain in the acceptable range. A high IgG4+/IgG+ percentage alone is also not sufficient for a diagnosis because in cases with a few plasma cells, an erroneously high ratio may be calculated owing to the relative proportions of both IgG and IgG4+ plasma cells. We are in agreement with other investigators with regard to the cut-offs for the absolute number of IgG4+ cells >50/high-power fields (HPF) and ratio of IgG4+/IgG+ cells >40%.31 We also propose selecting areas with the maximum density of IgG4+ plasma cells. At least three HPFs should be counted, and the average number of IgG4+ plasma cells per HPF should be calculated to give the results. In borderline cases, a descriptive diagnosis with a comment indicating "increased IgG4+ plasma cells" should be rendered along with a disclaimer for the clinician requesting additional work-up or follow-up to determine if the lesion indeed represents IgG4-RSD.31 A raised serum IgG4 level is not mandatory for the diagnosis but may be of valuable assistance. Serum IgG4 titer often correlates with disease activity and the number of involved organs, but levels may also be normal.40 It is noteworthy that increased serum IgG4 has also been reported in a host of other diseases, including atopic dermatitis, parasitic infections, pemphigus vulgaris, pemphigus foliaceus, and pancreatic carcinoma.
Not every entity with increased IgG4+ plasma cells and a high IgG4/IgG ratio can be accepted as belonging to the spectrum of IgG4-RSD. Requirements such as concomitant morphologic features and an appropriate clinical context are mandatory for diagnosis. Autoimmune diseases include cases with an increased proportion of IgG4+ plasma cells but this does not qualify for inclusion as IgG4-RSD because the morphologic features and clinical findings do not correspond with the diagnostic criteria.
CONCLUSION
In the past decade, significant progress has been made in our understanding of the genetic and immunologic triggers contributing to IgG4-RSD in addition to the discovery of the vast clinicopathologic spectrum of this entity. Our knowledge is currently evolving as newer developments come to light revealing a widely expanding histopathologic spectrum in this disease entity. Further studies are necessary to delineate the precise role played by IgG4 in the pathogenesis of this disease group in order to make meaningful contributions in the management of patients with this disease.





 XML Download
XML Download