

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Von Hippel-Lindau (VHL) disease is an autosomal dominant hereditary neoplasia syndrome that affects multiple organ systems.1,2 In VHL disease patients, renal cell carcinomas and cysts, pancreatic carcinomas and cysts, pheochromocytomas, and epididymal cystadenomas may develop.1,2 Moreover, central nervous system (CNS) manifestations, including CNS hemangioblastomas, retinal angiomatosis, and endolymphatic sac tumors, can occur.3,4 Among these tumors, CNS hemangioblastoma is one of the earliest features of VHL disease and has been found in 60 to 80% of patients with VHL disease.2-5 Spinal cord hemangioblastomas account for 13 to 50% of CNS hemangioblastomas associated with VHL disease and are a common cause of morbidity requiring treatment including surgical resection.1,2,4,6,7
Treatments of spinal cord hemangioblastomas have been published in many reports, and resection of these tumors is considered relatively safe with few complications.8-16 Thus symptomatic spinal cord hemangioblastomas should be treated with resection, even in VHL disease patients. However, the treatment strategy for asymptomatic spinal cord hemangioblastomas in VHL disease patients remains undetermined. CNS hemangioblastomas in VHL disease are reported to exhibit a two-step growth pattern of rapid growth and arrested growth in many cases, but some tumors show continuous growth without a quiescent phase.7,17 In addition, local growth factors such as vascular endothelial growth factor, placental growth factor, platelet-derived growth factor and epidermal growth factor have been shown to be elevated in hemangioblastomas, leading to upregulation of cyst formation and angiogenesis.18-20 As a result, it is quite difficult to predict the growth of hemangioblastomas in patients with VHL disease or their risk for neurological deficits. Recently, a few focused studies concerning spinal cord hemangioblastomas in patients with VHL disease have been reported, but they conceived somewhat different opinions regarding treatment of asymptomatic spinal cord tumors.7,17,21-24 A few authors advocated that resection should be limited to symptomatic spinal cord tumors,22,23 while others concluded that selective resection of asymptomatic tumors might be associated with better outcomes.17,21,24
In this regard, we reviewed spinal cord hemangioblastomas in VHL disease patients who had been treated at our institute. To devise an appropriate treatment plan for asymptomatic tumors, we compared asymptomatic tumors that had been resected to asymptomatic tumors that were not resected, as well as to symptomatic tumors.
MATERIALS AND METHODS
Data collection & patient population
We identified VHL disease patients who were surgically treated for spinal cord hemangioblastomas between 1999 and 2009. We reviewed all medical records and imaging studies from the initial diagnosis of VHL disease to the last visit. Patients who were followed for less than 12 months or patients with inadequate data for thorough evaluation were excluded. We also excluded patients whose conditions had deteriorated obviously because of brain tumors or other systemic problems regardless of spinal cord hemangioblastomas.
Patient evaluation
Symptoms, neurologic examinations, and functional status were serially recorded at the time of initial diagnosis, immediately before and after surgery, and at the last follow-up visit. All operative findings and surgical complications were also noted. Functional status of the patients was graded according to the scale proposed by McCormick, et al.25 MRI was performed for initial diagnosis, before surgery when necessary, shortly after surgery, when changes in symptoms or neurologic function occurred, and annually if patients were clinically stable. The number, location, and volume of tumors were measured using contrast-enhanced T1 weighted images, and cysts or syringes were evaluated from T2 weighted images. The relationship between tumors and the spinal cord was evaluated by the operation records. Tumor volume was calculated by the following formula: (length×width×height)×0.5.26 The sizes of cysts or syringes were quantified according to the number of vertebral columns matched with the involved spinal cord.
Surgical treatment
All operations were performed using the usual posterior approach. Patients were placed in the prone position. Laminectomies or laminotomies were performed beyond the cranial and caudal margins of the tumor for wide exposure of tumors. A midline dural incision was made. Dura mater was reflected laterally and retained with "stay-sutures". After gentle removal of arachnoid membrane from the surfaces of the tumor, supplying or draining vessels and crossing vessels were coagulated with bipolar cautery at their junctions with the tumor. Meticulous dissection between the tumor capsule and normal spinal cord was performed using micro dissectors and micro scissors. All supplying or draining vessels connected to the tumor were coagulated during dissection. The dissected plane was retained with small strips of Cottonoid, and circumferential dissection was performed. After complete dissection of the tumor, including the deep surface of the tumor, the tumor was removed en bloc. During the surgical procedures, motor evoked potential was monitored. Besides tumor resection, no other procedure was performed for syringes. Pre-operative angiography was performed in one case of a large tumor with a volume of 2400 mm3 from the cranio-cervical junction to C2. In this case, angiography was performed to identify the relationship between tumor vessels and the vertebral arteries.
Tumor grouping
To evaluate the influence of each tumor on the patient's clinical course, tumors were divided into three groups as follows: Group 1, tumors that were asymptomatic at initial diagnosis and were observed without resection; Group 2, tumors that were asymptomatic at initial diagnosis that were subsequently resected; and Group 3, tumors that were symptomatic at initial diagnosis and resected thereafter.
Tumors in Group 2 were resected as they were considered to exhibit a strong likelihood of leading to neurologic symptoms or deficits because of the very large size of the tumors or extensive syringes combined with the tumors. Similarly, surgical resection for a few tumors in Group 1 was performed when their size significantly increased during follow-up. However, there was no exact threshold for resection.
Statistical analyses
Non-continuous variables such as tumor location or the relationship between the spinal cord and the tumor were tested by Pearson's chi-square test and the linear-by-linear association method, and continuous variables were evaluated by the independent t-test (non-parametric Mann-Whitney test was used if needed). We used SPSS software (version 12.0.0, SPSS Inc., Chicago, IL, USA) for calculation. Statistical significance was determined by a two-tailed probability value of less than 0.05.
RESULTS
Patient demographics
Twelve patients (9 men, 3 women) fulfilled the inclusion criteria, and their mean age at the onset was 42.3 years (22-82 years). The average follow-up period was 49.3 months (12-130 months). Six of them had only one spinal cord hemangioblastoma, two patients had two tumors, two had three tumors, and two had four tumors (Table 1). Seven patients had intracranial hemangioblastomas and two had retinal capillary hemangioblastomas. Renal cysts were diagnosed in three patients, and pancreatic cysts were diagnosed in two patients. One patient presented with renal cell carcinoma. As mentioned previously, patients whose condition had deteriorated because of brain tumors or other systemic problems were excluded in this study. Thus, all accompanied manifestations listed above were in a well-controlled status (Table 1).
Tumor characteristics
We identified 24 spinal cord hemangioblastomas in 12 patients. Characteristics of these tumors are summarized in Table 2. The mean volume was 878.0 mm3 (18-5184 mm3). Six tumors were located in the cervical region, 14 in the thoracic region, and four in the lumbar region. There was no pure intramedullary tumor that required myelotomy, and 16 primarily intramedullary tumors were identified. Eight tumors were extramedullary tumors, in which more than half of the tumor burden was outside the spinal cord or located below the conus medullaris. All tumors were seated posterior to the dentate ligament. Cysts or syringes were accompanied in eight tumors. Seven tumors exhibited symptoms when diagnosed, and 17 did not. Among these 17 tumors, nine tumors (53%) were ultimately resected.
Comparison of asymptomatic patients and symptomatic patients
Five patients exhibited no symptoms or neurological deficits due to spinal cord hemangioblastoma when the tumors were initially diagnosed (Table 3). They remained stable after surgery, and were categorized as McCormick's grade I at the last follow-up. Three of the seven symptomatic patients were McCormick grade I, three were grade II, and one was grade III, initially. Among these patients, three patients showed a 1-point reduction in functional status at last follow-up, and one patient's condition extremely deteriorated from grade I to grade IV. Improvement was not observed for any patient (Table 3). Initial functional status was correlated with the final functional outcomes (p=0.026, data not shown). Moreover, the outcomes of the symptomatic patients were significantly worse than those of the asymptomatic patients (p=0.015) (Table 3).
Comparison of tumor groups
When we compare symptomatic tumors to asymptomatic tumors at initial diagnosis, asymptomatic tumors were significantly smaller (505.3 mm3 vs. 1787.4 mm3, p=0.043) and associated with a small syrinx (1.4 vertebral columns vs. 6.1 vertebral columns, p<0.001). Neither tumor location nor the tumor's relationship with the spinal cord was different between the symptomatic tumors and the asymptomatic tumors.
More specific results after dividing tumors into three groups according to the criteria mentioned above are shown in Table 4. There was no difference in age, tumor location, or relationship with the spinal cord among the three groups. Tumor volumes (p=0.018) and the sizes of combined syrinx (p=0.031) in Group 1 were significantly smaller than those in Group 2; similar results were also observed between Groups 1 and 3 (tumor volume; p=0.009, syrinx size; p<0.001). However, tumors of Group 2 were not significantly different in tumor volume (p=0.489) or syrinx size (p=0.072) from Group 3 tumors. Among 13 tumors in Group 1, nine tumors (69.2%) grew or developed symptoms during follow-up. The median progression-free survival of these tumors was 14.0±4.9 months. Statistical analysis for risk factors associated with progression-free survival was impossible due to the small number of tumors. Of the nine tumors that showed disease-progression, five tumors were resected. Two tumors were surgically resected because they caused neurologic symptoms. In one of them, the neurologic deficit was not improved after surgery, and the functional status of the patient deteriorated from McCormick grade I to grade IV. Three tumors were resected as they grew large enough to be considered as posing a potential risk of causing neurologic deficits. The other eight tumors in Group 1 did not produce any symptoms or deficits up to the last follow-up visits, and showed no or minimal growth. In Group 2, no newly developed neurological symptom or deficit was seen after surgery, and all patients were stable during the follow-up period. In five tumors of Group 3, the neurologic symptoms and deficits were permanent. In three of these five tumors, the McCormick scale dropped one grade because of recurrence or as a result of surgical procedures associated neural injury.
Review of individual asymptomatic tumors with resection
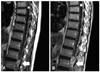
Information on the asymptomatic tumors resected during follow-up is summarized in Table 5. The patient with tumor No.1 (Fig. 1) presented with pain in the lower extremities and difficulty with voiding. He received surgery for another tumor responsible for his symptoms, located at L3-5. After surgery, his neurologic symptoms and deficits disappeared. Although tumor No.1 at L1-2 was large, it was not resected because the patient had two additional spinal cord tumors and showed extensive leptomeningeal enhancement along the spinal cord on MRI (Fig. 1A). On an MRI taken 15-months after the surgery (8-months after the last MRI study), the tumor increased almost twice in size (Fig. 1B). We decided to perform an operation thereon, but after several days between the outpatient visit and admission, paraparesis occurred and rapidly progressed. Moreover, he became stuporous before surgery because of recurrence of the cerebellar hemangioblastoma and associated acute hydrocephalus. After resection of the cerebellar hemangioblastoma and several days of conservative care, the patient fully woke up. However, motor function in his legs did not return (motor grade III) and was not improved after the spinal tumor resection. Tumors No.2 and No.3 in the same patient with the cerebellar hemangioblastoma were identified while screening his spinal MRI (Tumor No.2 is illustrated in Fig. 2). The two tumors were observed without resection for a long time because their growth was slow. When they grew to a relatively risky volume of 468 mm3 and 245 mm3, respectively, they were simultaneously resected, at 45 months after the initial diagnosis. The patient with tumors No.4 and No.5 was planned to be observed. However, in spite of small tumor size, tumor No.4 on T12 and the accompanied syrinx caused difficulty with voiding and mild myelopathy, so the operation was performed one month after the initial diagnosis. After surgery, the symptoms were improved. Tumor No.5 on T1-2 was resected at the same time. Tumors No.2 through 5 caused no newly-developed neurological symptoms or deficits during the period of the immediate post-operative time to their last follow-up visit, and the patients' functional status was evaluated as McCormick's grade I. As previously noted, four tumors in Group 2 (No.6-9 in Table 5) were safely resected without any new neurologic symptom, deficit or surgical complication.
DISCUSSION
VHL disease is a multi-organ familial neoplasia syndrome caused by a germline mutation in the VHL tumor suppressor gene,27 and it is transmitted in an autosomal dominant fashion with greater than 90% penetrance.28 In many visceral or CNS lesions associated with VHL disease, spinal cord hemangioblastomas are the most common cause of morbidity,7 and accordingly, should be cautiously treated. However, the outcomes of spinal cord hemangioblastomas in VHL disease patients seemed to be poorer than those of sporadic spinal cord hemangioblastomas.5 Thus, it is very important to identify the factors affecting the outcomes of VHL patients with spinal cord hemangioblastomas and to establish a proper treatment strategy.
In previous reports, pre-operative neurological status, ventral tumor location, and tumor volume were presented to be correlated with the functional outcomes of the patients. However, there were some differences in these regards among the authors. Kanno, et al.21 reported that the surgical outcomes for tumor volume <500 mm3 (10 mm in diameter) were better than those >500 mm3, while Lonser, et al.22 showed that patients with no or minimal preoperative neurological dysfunction, lesion size smaller than 500 mm3, and with dorsal lesions were more likely to have no or minimal neurological impairment. Ventral tumors or completely intramedullary tumors were associated with an increased risk of post-operative worsening in Mehta, et al.'s study.23 Additionally, Ammerman, et al.17 showed that tumor size was the only variable predictor for the development of symptoms and eventual need for therapy. Based on these results, different treatment plans have been proposed. Van Velthoven, et al.24 concluded that spinal cord hemangioblastomas should be resected when they progress on radiologic studies. However, Lonser, et al.22 and Mehta, et al.23 advocated timely selective removal of only symptomatic tumors, because CNS hemangioblastomas frequently show a two-step pattern of growth consisting of a growth phase and a quiescent phase. In addition, Kanno, et al.21 suggested that surgical treatment should be considered before the tumor volume exceeds 500 mm3 on MRI during follow-up. Ammerman, et al.17 reported that surgical treatment of asymptomatic tumors might be associated with less risk and improved neurological outcomes when the tumors are smaller and when surgical treatment is performed before tumors cause neurologic deficits, if they are predicted to produce symptoms and eventually require treatment within the next few years. They also recommended the combined volume of the tumor and cyst to be used as a predictive marker for the development of symptoms and eventual need for therapy.
In this study, five asymptomatic patients and seven symptomatic patients were markedly different in their final functional outcomes, and the outcomes were firmly correlated with initial neurological status. Also, neurological symptoms and deficits were shown to be affected by tumor volume and the extent of the combined syrinx. In other words, VHL patients with a large tumor and an extensive syrinx were at a greater risk of neurological deficits, some of which may be irreversible. Accordingly, asymptomatic spinal cord hemangioblastomas that posed a greater risk of causing neurological deficits were resected at the time of initial diagnosis at our institute, and they were evaluated in this study as Group 2 tumors. These tumors were as large as symptomatic tumors (Group 3), whereas the corresponding patients (Group 2) showed better outcomes. Therefore, it was thought reasonable to conclude that the functional outcomes of patients with a large tumor are affected more by the presence of neurological symptoms and deficits than by the tumor volume itself, and thus resection of significantly large asymptomatic tumors might bring about better outcomes.
Based on the results of this study and previous reports, we recommend selective surgery for asymptomatic spinal cord hemangioblastomas in VHL disease before the tumors cause neurologic symptoms. Our proposal for the treatment of spinal cord hemangioblastomas in VHL disease is illustrated in Fig. 3. Large tumors are strongly associated with the generation of symptoms, and a tumor volume of >500 mm3 is regarded to be correlated with poor functional outcomes.17,21,22 Thus, tumors >500 mm3 should be resected regardless of whether they are asymptomatic. Tumors smaller than a volume of 500 mm3 can be followed at approximately 1-year intervals. However, if symptoms or neurological deficits occur during follow-up, resection must be performed. It was previously shown that 98% of the spinal cord hemangioblastomas larger than a volume of 51 mm3 (4.7 mm in diameter) required surgery in a more than 10-year long-term follow-up study.17 Also, syringes were shown to be associated with the development of neurological symptoms and deficits in our study and by Wanebo, et al.7 In addition, the growth rates of spinal cord hemangioblastomas associated with syringes were reported to be much higher than those of spinal cord hemangioblastomas not associated with syringes, and the mean growth rate of spinal cord hemangioblastomas associated with syringes was about 50 mm3/month.7 Therefore, progressively growing tumors larger than a volume of 51 mm3 in combination with syringes are predicted to produce neurological symptoms or deficits within several months to a year, and resection of these tumors might result in better outcomes when surgical treatment is performed before they cause neurological symptoms or deficits. Therefore, we recommend considering surgery for these tumors or intense observation at short intervals of 6 months or less (Fig. 3). In conclusion, the clinical features of VHL disease are very complicated, and VHL disease patients should be thoroughly evaluated. Moreover, spinal cord hemangioblastomas without symptoms must be closely observed, and, in particular, if they are significantly large or combined with an extensive syrinx, surgical resection should be performed before neurological deficits occur.




 XML Download
XML Download