

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Centronuclear myopathy is a group of pathologically defined disorders that characteristically have a high proportion of small myofibers with centrally placed nuclei.1 Spiro, et al.2 first described the term "myotubular myopathy" in 1967. A severe form, called X-linked myotubular myopathy (XLMTM), is related to severe clinical features, such as hypotonia, generalized weakness, feeding difficulty, and respiratory distress requiring ventilator support. Patients with XLMTM show typical dysmorphic features characterized by severe hypotonia, generalized muscle weakness, and impaired maturation of muscle fibers, causing perinatal death of male infants.3 XLMTM pathogenesis is due to a mutation in the MTM1 gene on Xq28;4,5 it encodes a phosphoinositide lipid phosphatase, which is known as myotubularin and appears to be important in muscle maintenance.6,7
To our best knowledge, this is the first report on an XLMTM family with 2 affected infants diagnosed by muscle biopsy and gene analysis in Korea. A nonsense mutation Arg486STOP was identified in exon 7 of the MTM1 gene in this family.
CASE REPORT
Patient 1 (IV 4)
A preterm male was born at 34 weeks of gestational age by normal spontaneous vaginal delivery and had a birth weight of 2.41 kg. Apgar score was 2 at 1 min, 6 at 5 min, and 7 at 10 min. He had generalized muscle hypotonia that required ventilation and a lack of spontaneous movement. His mother, who underwent spontaneous abortion prior to this pregnancy, had a healthy daughter. During the antenatal period, severe hydramnios was detected. She had a family history of a cousin who was diagnosed with muscle disease and expired at the age of 3. Patient 1 was admitted to the Neonatal Intensive Care Unit after birth, and there were no signs of spontaneous movement or respiration. He showed a small, thin rib cage on the chest radiography. Lactate dehydrogenase (LDH) 2286 was elevated only. Tandem mass spectrometry screenings including amino acids, organic acids, fatty acids metabolic disorders, brain ultrasonogram, and electromyography were non-specific. Although he was managed conservatively, the hypotonia was not improved. Therefore, we performed a chromosome analysis, gene analysis, and muscle biopsy to rule out myopathy. Results showed Arg486STOP genetic sequence and specific myotubular myopathy in muscle histology. He was confirmed as XLMTM. Self respiration and spontaneous movement were not present after 55 days of life, therefore, his parents decided to withdraw care.
Patient 2 (IV 5)
The mother of the above case did not follow up at our hospital in spite of our strong recommendation. Three years later, however, the mother visited us at the time of her third delivery. She had severe hydramnios similar to her previous pregnancy. A male baby was born at 36 weeks of gestation by cesarian section. He also showed severe hypotonia and respiration difficulty. A ventilator was used, and conservative care was given. The clinical courses were similar to those of patient 1.
Muscle biopsy and gene analysis for the MTM1 gene were performed and confirmed the diagnosis of XLMTM. He was discharged and expired 28 days after birth. The entire family, including the mother and healthy daughter, visited our genetics clinic to undergo gene analysis for the MTM1 gene. The gene analysis revealed that the mother was a carrier of XLMTM, but the daughter was not (Fig. 1).
Muscle Biopsy
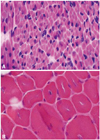
Histological findings of the muscle biopsy were consistent with centritubular (myotubular) myopathy. HE-staining showed central nuclei in very high proportions of the relatively small myofibers. In addition, severely hypoplastic muscle fibers with centrally placed nuclei were observed (Fig. 2).
Genetics
In view of the morphological findings, an analysis for the MTM1 gene was performed and Arg486STOP, a nonsense mutation, was detected (Fig. 1).
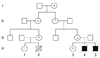
Pedigree (Fig. 3)
This pedigree is based from family history. The son of mother's first cousin died from an unknown myopathy at 3 years old. He had been bed rest with home ventilator till that time.
DISCUSSION
XLMTM is a rare disease that affects one out of 50,000 males and is due to MTM1 mutations.6,8 This disease is caused by a maturational arrest of the fetal muscle during the myotubular stage of development from the 8th to 15th week of gestation. Compared to other congenital myopathies, patients with XLMTM have a rather homogeneous clinical presentation. They are characterized by severe hypotonia, generalized muscle weakness, impaired maturation of muscle fibers, and dysmorphic features associated with weakness. Antepartum symptoms of XLMTM include polyhydramnios (the most common feature), poor fetal movement, and fetal cardiac arrhythmias. A high percentage of these patients require mechanical ventilation from birth throughout the duration of life.9 Many patients show sucking/swallowing impairments, ophthalmoplegia, facial weakness, and joint contractures. They are rarely able to walk and remain severely hypotonic.
About 50% of patients who survived for 1 year need 24-hour ventilatory support, and approximately 75% of severely affected neonates die within the first few weeks or months of life due to respiratory insufficiency.8 The average life expectancy of XLMTM patients is 29 months, but a number of papers reported adult survivors of the disease.10-12 A female carrier of XLMTM is generally asymptomatic and slowly pregressive, and the mortality is not high compared to affected males.13,14
To confirm XLMTM, a histological examination of muscle specimen is necessary. The high proportion of centrally placed, relatively large nuclei on tinctorial stains are the typical findings of XLMTM.3 Gene analysis for MTM1 gene mutations is necessary to confirm XLMTM, however, some patients with mild XLMTM are found to have no centrally located nuclei in muscle biopsies.15
The gene MTM1, which is linked to XLMTM, is well established.16 The MTM1 gene is located on the proximal segment of the long arm of X chromosome (Xq28),17 and it consists of 15 exons and has an encoding sequence of myotubularin with a tyrosine phosphatase domain. The mutations are widespread, and are found in exons 4, 12, 3, 8, 9, and 11. Truncating mutations are the most common point mutations of XLMTM associated with a severe and early lethal phenotype, while some missense mutations are the cause of relatively milder forms of XLMTM; such as autosomal dominant centronuclear myopathy (ADCNM) that present slowly in adults with a diffuse weakness and autosomal recessive centronuclear myopathy (ARCNM) that presents muscle weakness in infancy or early childhood. The mutation in our cases resulted from an Arg486STOP nonsense mutation, and the mother of the patients was revealed to be the carrier of the mutation.
To our best knowledge, this is the first case of sibling infants diagnosed with XLMTM by muscle biopsy and gene analysis. The descriptions of the two patients with severe XLMTM from the same mother provide an important lesson for clinicians to evaluate infantile muscular disorder (whether it saves the baby's life or not) and to consult with the mother about prenatal family planning and antenatal genetic screening.18 Despite the fact that pedigree analysis (Fig. 3) provides the first sign of a de novo mutation, gene studies on the healthy daughter in the XLMTM family are necessary because she may be a carrier of the XLMTM mutation. This will greatly affect her future if she decides to have her own children.14

 XML Download
XML Download