

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Pheochromocytomas are tumors that arise from chromaffin tissue. While most pheochromocytomas arise usually in the adrenal medulla, there are also extra-adrenal pheochromocytomas (paragangliomas) that arise in an extra-adrenal site along the sympathetic or parasympathetic chain.1 Pheochromocytomas produce and secrete catecholamines that cause a variety of clinical symptoms, including headache, sweating, paroxysmal or sustained hypertension, and heart palpitations. These symptoms can result in a decreased quality of life.
Pheochromocytomas are relatively rare tumors found in approximately 4% of adrenal incidentalomas.2 However, the number of tertiary center referrals for pheochromocytomas are reportedly increasing, likely as a result of improved detection.3 Due to the rarity of this disease, there have not been many large-scale studies regarding its clinical characteristics. Goldstein, et al.4 reported 104 patients with pheochromocytoma over a period of 48 years, and Mannelli, et al.5 reported a multicentric study of 284 patients with pheochromocytoma. In Korea, Lee and Oh6 reported clinical evaluation of pheochromocytoma in only 12 patients in 1993. Other studies regarding pheochromocytoma are primarily case reports.
We investigated the medical records of 119 patients with pheochromocytoma at our institute over the last 23 years and analyzed their epidemiological, clinical, laboratory, radiological, and pathological data.
MATERIALS AND METHODS
From 1986 to 2009, 119 patients were diagnosed with pheochromocytoma at our institute, and all were included in our study. All patients underwent surgical excision of their tumor, and the diagnosis was confirmed by pathological findings in all cases. Records of epidemiological, clinical, laboratory, radiological, and pathological data were retrospectively reviewed and analyzed.
We investigated the presence of associated symptoms including anxiety, visual blurring, palpitations, headache, sweating, and abdominal pain. We also investigated whether patients presented with associated symptoms or if an adrenal mass was incidentally found during a routine medical exam or imaging study. We also investigated the presence of persistent hypertension or diabetes and any possible association with familial pheochromocytoma syndromes.
Laboratory studies included 24-hour measurements of total urinary catecholamines, epinephrine, metanephrine, norepinephrine, dopamine, vanillylmandelic acid, and plasma metanephrine, norepinephrine, and epinephrine. Laboratory studies were considered abnormal in cases where these values were elevated.
Tumor location and dimensions were obtained from radiologic studies, including computed tomography (CT), magnetic resonance imaging, and positron emission tomography. In cases where iodine-131-labeled metaiodobenzylguanadine (131I-MIBG) scintigraphy was performed, the results were compared with the pathologic findings.
The diagnosis of malignancy was based on the presence of metastasis. In patients who were diagnosed with malignant pheochromocytoma, the time of diagnosis and the treatment of the malignancy were reviewed. If the patient was deceased, the time from diagnosis to death was also noted.
RESULTS
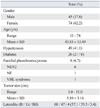
Of 119 patients, 45 were male and 74 were female, and mean age at surgery was 43.83 ± 13.49 (11-74) years (Table 1). Symptoms associated with pheochromocytoma were documented in 99 patients (83.2%). However, 43 patients (36.1%) were diagnosed with pheochromocytoma incidentally during a routine medical exam or during imaging studies for unrelated disorders, regardless of the presence of symptoms. Of 43 patients, ten cases occurred prior to 2000. Persistent hypertension was present in 49 patients (41.1%), and diabetes was present in 26 patients (21.8%). Sporadic cases of pheochromocytoma accounted for 111 patients (93.3%), while eight patients (6.7%) were associated with familial syndromes. Of these eight cases, six were multiple endocrine neoplasia type 2 (MEN2), one case was neurofibromatosis (NF), and one case was von Hippel-Lindau (VHL) syndrome. The mean age of the patients with familial syndromes was 39.88 ± 14.37 (25-71) years.
Fifteen patients showed normal laboratory values. Ten of these patients underwent preoperative management for suspected pheochromocytoma, while the other five underwent surgery without preoperative management. Three of these patients exhibited severe changes in blood pressure during surgery. The most commonly used medication for preoperative treatment was phenoxybenzamine, which was utilized in 90 patients (75.6%). Doxazosin was used in 21 patients (17.6%), and prazosin was used in three patients (2.5%). In 6 patients (5%), beta-blockers were administered in addition to an alpha-blocker. The length of preoperative management was variable with a range of two to eight weeks.
The mean dimension of the tumors, according to radiologic studies, was 5.89 ± 3.18 (1.0-15.0) cm. Forty-seven tumors (39.5%) were left-sided, 68 (57.1%) were right-sided, and four (3.4%) were bilateral. Of the 4 patients with bilateral tumors, two were associated with MEN2, and one was associated with VHL syndrome. The other patient was diagnosed with malignant pheochromocytoma. 25 patients received 131I-MIBG scintigraphy. The study yielded false negative results in 6 patients, with two of these patients being asymptomatic and showing normal values in laboratory studies.
Of our 119 cases of pheochromocytoma, 43 patients (36.1%) underwent laparoscopic surgery. Laparoscopic surgery was first performed at our institute in 1990, and the first laparoscopic adrenalectomy was performed in 1996. Forty of the 43 cases of laparoscopic adrenalectomy were performed after 2000.
8 patients were diagnosed with malignant pheochromocytoma, which was established in the presence of distant metastasis either at the time of diagnosis or during follow-up (Table 2). The mean dimension of the malignant tumors was 7.81 ± 4.01 (3.0-15.0) cm, and the mean age of these patients was 49.63 ± 11.94 (28-71) years. In 1 patient, a metastatic lymph node in the neck was found at the time of diagnosis of pheochromocytoma. The patient underwent surgical excision of the tumor and radiotherapy for bone metastasis; the patient subsequently died of malignant pheochromocytoma 27 months after diagnosis. 7 patients were diagnosed with malignant pheochromocytoma at a mean of 49 ± 25.83 (6-75) months after surgery because of metastases to the liver, lung, bone, or lymph nodes. Of these seven patients, five died of malignant pheochromocytoma. The mean time to death for the 6 patients who died of malignant pheochromocytoma was 31 ± 28.71 (1-81) months. 2 patients received a regimen of cyclophosphamide, vincristine, and dacarbazine (CVD) chemotherapy; one of these patients received radiotherapy also for bone metastasis. Of the three patients who received conservative care, one died one month after treatment, one died seven months after treatment, and one patient died at 81 months after treatment. Two patients survived for 15 and 24 months, respectively, into the follow-up period. One patient underwent 131I-MIBG treatment for lung metastasis, and the other patient underwent surgical excision of lung and lymph node metastases.
DISCUSSION
In the present study, we reviewed the records of 119 patients with pheochromocytoma and confirmed variable clinical features of the disease. Hypertension, whether sustained or paroxysmal, is the most common clinical sign, and headache, excessive truncal sweating, and palpitations are the most common symptoms.7 In a retrospective study of 41 patients with pheochromocytoma, blood pressure anomalies led to the discovery of pheochromocytoma in only 51% of patients, and only 59% of patients suffered from hypertension. In our review, 47 patients (41.1%) were found to have suffered from persistent hypertension, and 99 patients (83.2%) presented with symptoms associated with pheochromocytoma. Pheochromocytoma was found incidentally in 43 patients (36.1%), only ten of whom were diagnosed prior to 2000. Kopetschke, et al.8 reported that approximately 30% of pheochromocytomas are detected incidentally, and that this proportion is increasing. This increase in detection is likely due to increasing availability of imaging procedures.
Diabetes is also frequently found in patients with pheochromocytoma and is related to catecholamine-induced insulin resistance and insulin suppression.9 The reported incidence of diabetes in patients with pheochromocytoma varies from 15.5% to 48.3%.10-13 In young patients with hypertension and a normal body weight, the presence of diabetes is a clinical predictor of pheochromocytoma.9 However, the symptoms and signs of pheochromocytoma are variable and non-specific, therefore, clinical detection of pheochromocytoma is very difficult, and diagnosis is sometimes delayed or missed completely.5,14 This may explain relatively high prevalence of pheochromocytoma discovered at autopsy, approximately 0.05%.15,16
It is widely believed that approximately 10% of pheochromocytomas are associated with familial syndromes, however, it is now recognized that the frequency of germline mutations in apparently sporadic presentations is as high as 15% to 24%.1 Sporadic pheochromocytoma is usually diagnosed in individuals between 40 and 50 years of age, whereas hereditary forms are diagnosed earlier, most often before the age of 40.14 The probability of developing bilateral or multifocal tumors is higher in hereditary forms.17 In the present study, 75% (3/4) of bilateral tumors were associated with familial pheochromocytoma syndromes, and a CT image of a patient with VHL syndrome showed multiple and bilateral tumors. Germline mutations in six genes are related to familial pheochromocytoma. Prior to 2000, it was thought that familial pheochromocytoma was caused by germline-inactivating mutations in the RET proto-oncogene for MEN2, mutations in the tumor suppressor gene VHL, and mutations in the NF1 genes. More recently, the succinate dehydrogenase D, B, and C (SDHD, SDHB, SDHC) genes, which are associated with familial pheochromocytoma and/or paraganglioma syndrome, have been identifed.14 Of these six genes, SDHB mutations are associated strongly with extra-adrenal and malignant pheochromocytoma.18-20 Whether or not genetic testing should be performed in all patients with sporadic pheochromocytoma remains controversial. In a previous study, approximately one-fourth of patients with pheochromocytoma were found to have mutations, and some authors have suggested that genetic testing should be offered to all patients.21,22 However, Pigny, et al.22 suggested that genetic testing should be performed in patients who were diagnosed before age 20 (prevalence of hereditary forms: 33.3%) or who had a bilateral pheochromocytoma (prevalence: 75%) and should be strongly recommended in patients diagnosed prior to age 50 (cumulative prevalence: 10.4%). They did not recommend genetic testing in patients with a unilateral pheochromocytoma diagnosed after 50 years of age. In our study, only eight patients (6.7%) were associated with familial syndromes. Four of these patients were found to have MEN2 and were diagnosed with pheochromocytoma only after surgery for medullary thyroid cancer; genetic testing was not performed in the majority of these patients.
The diagnosis of pheochromocytoma is usually based on biochemical testing in suspected patients or in patients with an incidentaloma. Biochemical evidence of excessive catecholamine production is an essential step in the diagnosis of pheochromocytoma. However, many physiologic stimuli, drugs, and clinical conditions can produce false-positive results. Furthermore, pheochromocytomas do not always secrete enough catecholamines to produce positive test results and often secrete catecholamines only episodically.7 Missing the diagnosis of pheochromocytoma can lead to catastrophic consequences. In this study, fifteen patients (13%) demonstrated normal values in laboratory testing. Five of these patients did not have clinical symptoms or signs suggestive of pheochromocytoma, and therefore, preoperative management was not performed. Three patients demonstrated severe changes in blood pressure during surgery. In order to establish an accurate diagnosis, it is advisable to perform two independent urinary collections to account for the episodic nature of pheochromocytoma.1 Unfortunately, laboratory studies were not performed twice in the 15 patients who had false-negative results. Measurement of the plasma levels of metanephrine is more sensitive than those of other biochemical tests.1,7 The sensitivity of plasma metanephrine level is nearly 99%, and the specificity has been reported to range from 85% to 89%.23,24 Furthermore, a plasma metanephrine measurement is also sufficient to exclude pheochromocytoma because of its high negative-predictive value.25 Thus, many authors recommend to obtain a plasma metanephrine measurement as an initial biochemical test.1,7,23
The World Health Organization's definition of a malignant pheochromocytoma is the presence of metastasis; the diagnosis of malignancy can be made only retrospectively when metastases have become evident. Metastases occur most frequently in the bone, liver, lungs, and lymph nodes, and can appear as many as 20 years after initial presentation.1 Clinical, biochemical, and radiological features are inadequate to either predict malignancy or to distinguish benign from malignant lesions. Local invasion and various histopathological features can be suggestive of malignancy, however, these features are not widely accepted, and more sensitive and specific diagnostic means need to be developed.17 In general, tumors that are large (> 5 cm) or have an extra-adrenal location have a higher risk for malignant disease, and paraganglioma in patients with SDHB gene mutations have a particularly high rate of malignant disease.14 The overall five-year survival in patients with malignant pheochromocytoma ranges from 40% to 74%.17 At present, there is no effective treatment for malignant pheochromocytoma. Treatment with 131I-MIBG or combination chemotherapy (cyclophosphamide, vincristine, and dacarbazine) has produced disappointing results, producing only short-lived remissions.14 Recently, attempts have been made to create targeted therapies interfering with specific pathways within pheochromocytoma cells. Park, et al.26 reported a case of malignant pheochromocytoma that demonstrated a good metabolic response to sunitinib.
Tumor recurrence has been reported in up to 17% of patients in a large study.27 There are no reliable tests available to distinguish benign from malignant pheochromocytomas. Thus, all patients should be followed-up indefinitely, and long-term clinical and biochemical assessments are necessary. In this study, the proportion of patients with familial pheochromocytoma might have been underestimated because genetic testing was not performed in most patients. However, genetic testing should be considered in all patients, especially in patients with a family history, young age, and/or multifocal, bilateral, extra-adrenal, or malignant tumors.

 XML Download
XML Download