

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
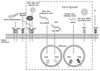
Viruses have many epitopes to induce immune responses from both innate and adaptive immune systems. Especially, viral double-stranded RNA (dsRNA), single-stranded RNA (ssRNA), unmethylated CpG motif and outer structural proteins stimulate intrinsic cellular defenses and further innate immune response1-5 (Fig. 1). These viral ligands are recognized by Toll-like receptor (TLR) during the virus replication. TLR is one of the most common pattern recognition receptor (PRR) of intrinsic cellular defenses that recognize pathogen-associated molecular patterns (PAMPs) such as bacterial or viral components. TLRs are composed of three general components, such as extracellular domains (ECDs), transmembrane domains, and cytoplasmic tails which contain Toll/interleukin-1 receptor (TIR) for signaling.6-8 TLR ECDs contain 19-27 leucine-rich repeats (LRRs) and cysteine-flanking regions, which form horseshoe-like structure to recognize each specific type of PAMPs for TLR. The TIR of cytoplasmic tail of TIR mediates downstream signal transductions.7,9 As a means for early recognition of microbial pathogens, TLRs are expressed on various cells,10 their recognition signals leading to induction of innate immune responses.
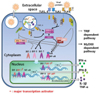
Recognition of these various viral components is possible because of their N-terminal ECD which contain 18-25 tandem copies of a short (24 residues) motives which are known as the LRR.5 Up to date, 13 human TLRs are discovered, and most of their functions have been elucidated. There are 5 major sensors for viral infections, such as TLR2, TLR3, TLR4, TLR7, TLR8 and TLR9. TLR3 recognizes viral dsRNA, TLR2 and TLR4 detect viral structural proteins or glycoproteins, TLR7 and TLR8 recognize viral ssRNA, and TLR9 senses viral CpG motif during replication of many viruses (Table 1). Therefore, these TLR constitute a powerful sensor system to detect viral components.11 In fact, each TLR has different signal transduction pathway, either MyD88-dependent or TIR domain-containing adaptor inducing interferons (IFNs)-β (TRIF)-dependent. Only TLR3 uses TRIF-dependent pathway (Fig. 2), whereas TLR7, TLR8 and TLR9 use MyD88-dependent pathway. On the other hand, TLR4 particularly uses both TRIF-dependent and MyD88-dependent pathways. Lower steps of these pathways, including transcription activators such as interferon regulatory factor (IRF)-3, 5, 7 and nuclear factor-κB (NF-κB), are activated. The NF-κB is one of the major transcription factors that regulates various innate immune responses, such as inflammation.12-15 The IRF families are known to induce type I IFNs,16,17 and type I IFNs secreted make individual cells to be resistant against viral infection. Focusing on antiviral activities, these activated transcription factors are essential for induction of type I IFNs, especially IFN-α and IFN-β.11 There are several isotypes of type I interferon, such as IFN-α, IFN-β, IFN-δ, IFN-ω and IFN-τ.18 Especially, IFN-α and IFN-β have been shown to be potent antiviral cytokines. They not only inhibit viral replication directly, but also activate immune effector cells.18 In innate immunity, type I IFNs have been demonstrated to play a crucial role in dendritic cell maturation, differentiation, B cell activation, priming of primary antibody responses, and memory CD8+ T cell proliferation, and to prolong long-term survival.19 Moreover, IFN-α/β induce GTPase-like myxovirus-resistance protein (Mx) protein which inhibits virus replication.20 Through positive feedback processes, IFN-α is able to enhance many TLRs, such as TLR3, TLR4, TLR7, and TLR8.11 These aspects of viral replication and invasion are reviewed herein.
TOLL-LIKE RECEPTORS SENSING VIRAL LIGANDS
Almost all viruses are composed of nucleic acid and viral proteins. They are detected by specialized sensors such as TLRs, one of the PRR. TLRs are localized on the cell surface or endosomal membrane. Therefore, attachment of a virus is sensed by external TLRs, especially TLR2 and 4 on the cell surface, while invasion of a virus is recognized by internal TLRs (TLR3, 7, 8 and 9). Each step of viral replication produces many PAMPs, such as single- or double-strand RNA, viral nuclear proteins, envelope proteins and glycoproteins. Signaling from TLRs sensing can activate IRF3, 5, 7 or NF-κB pathway, resulting in induction of type I IFNs. The secreted type I IFNs make neighbor cells to express various antiviral proteins, such as myxovirus-resistance protein (Mx) GTPase, ribonuclease L (RNase L), RNA-dependent protein kinase (PKR), Oligo-adenylate synthetase (OAS), and Interferon stimulated gene (ISG).
TLR3
Double-stranded RNA (dsRNA), which is a replication intermediate of several viruses, sensitize innate immune system through TLR3. They are mostly enveloped RNA viruses like Respiratory Syncytial virus (RSV),21 influenza A virus,22 and West Nile virus (WNV).2 Double-stranded RNA is observed during most RNA virus replications, and is correlated with endosomal expression of TLR3. Role of TLR3 is experimentally proved using an artificial dsRNA and TLR3 knockout mice. Synthetic dsRNA polyinosinic-polycytidylic acid (poly(I:C)) has an immunostimulatory activity similar to dsRNA,23 and TLR3-deficient mice have been found to have their response to dsRNA impaired.15 This sensing ability against viral dsRNA is due to molecular structure of TLR3. Recently, TLR3-ECD has been shown to resemble a horseshoe-shaped solenoid of 23 LRRs, with each LRR forming one turn of the solenoid. As a dsRNA binding motif of TLR3, H539 and N541 are conserved in living organisms, ranging from zebrafish to humans: H539 coordinates a phosphate group, whereas N541 forms a hydrogen bond to the 2'OH of the ribose. When longer dsRNAs are recognized by TLR3, binding of TLR3s to longer dsRNAs forms oligomer morphology. This model can explain the mechanism of why most oligomer TLR3 easily accommodates the formation of a TIR dimers on the endosomal membrane;5 particularly, dsRNA has never been detected in non-infected cells, while detected only in virus-infected cells. Therefore, TLR3 is regarded as a major PRR against virus in animal cells. In fact, respiratory epithelial cells over-express TLR3 when infected by influenza A virus, and cells are then able to detect viruses' infection signals and able to acquire resistance.24
TLR2
TLR2 is located on the cell surface and recognizes extracellular ligands. TLR2 forms heterodimers with either TLR1 or TLR6. Several viruses, such as Herpes simplex virus (HSV), cytomegalovirus (CMV), hepatitis C virus (HCV) and measles virus, induce type I IFNs and proinflammatory cytokines via TLR2 signaling.25-29 HSV infection induces TLR2-mediated pro-inflammatory cytokines and type I IFNs production.26 It seems likely that viral outer glycoprotein acts as ligand for TLR2. However, HSV's four essential viral glycoproteins, such as glycoprotein B (gB), D (gD), H (gH), and L (gL), have recently been found to induce innate immune recognition in TLR2-independent manner.27 However, HSV's ligand for TLR2 is still largely unclear. CMV is also recognized by TLR2 and CD14. Recognition signal triggers inflammatory cytokines and type I IFNs production. Especially, TLR2 is required for NF-κB activation in response to CMV infection.28 Viral core protein and nonstructural protein (NS) 3 of HCV are potent ligands for TLR2. Amino acid sequences in the regions of 2-122 in core and 1450-1643 in NS3 are required for recognition by TLR2.29 Hemagglutinin (H) protein of wild type measles virus can induce proinflammatory cytokines such as interleukin (IL)-6 by TLR2 activation. Inactivated virus by heating or ultraviolet (UV) inactivation can also induce TLR2-dependent signaling. On the other hand, point mutation in H protein of measles virus affects TLR2 signaling.25 Moreover, TLR2 is able to induce intrinsic cellular defenses by sensing these viral ligands, resulting in the production of type I IFNs.
TLR4
Similar to TLR2, TLR4 is also located on the cell surface and forms heterodimers with either TLR5, which presumably enhances its activity, or TLR1, which inhibits its activity.3 Otherwise, MD-2 is essential for signaling of TLR4. N-linked carbohydrate seems to have critical role in interaction between MD-2 and TLR4. In human, MD-2 contains 2 N-glycosylated sites, while TLR4 contains 9 N-linked glycosylated sites. The site-directed mutagenesis studies show that these glycosylated sites are cross-linking positions between MD-2, TLR4 and its ligands.30 The major ligand of TLR4 is lipopolysaccharides (LPS) derived from the outer membrane of Gram-negative bacteria, however, there is also an evidence to indicate that TLR4 recognizes also some viral components, such as viral glycoproteins on viral particles. It is known that RSV infection induces up-regulation of TLR4 on airway epithelial cells,31 and a fusion (F) protein of RSV, one of the glycoproteins, sensitizes TLR4. Since, purified RSV F protein can activate MD-2-dependent TLR4 signaling pathway, therefore, combination of TLR4 with MD-2 appears to be indispensable for immune response against viruses.3 Furthermore, among the TLRs 1 through 13, only TLR4 showed an altered expression after HCV infection of human B cells. Even though HCV's agonist for TLR4 has not yet been clearly identified, it is certain that HCV has a ligand against TLR4. When a siRNA targeting TLR4 is applied, HCV-induced cytokine production is specifically inhibited.32 Indeed, HCV is used as an agonist for TLRs, especially TLR4.33 Also, mammary tumor virus (MMTV) induces TLR4-dependent innate immune responses, confirmed by using TLR4 knockout mice. There was a large amount of cytokine released from TLR4-sufficient mice, whereas these was not from TLR4 knockout mice.34 An envelope glycoprotein SU is regarded as a legend for TLR4,35 therefore, extracellular TLR4 seems to play a critical role in intrinsic cellular defense against virus infection.
TLR7 and TLR8
Both TLR7 and TLR8 sense the viral infection by recognizing uridine-rich single-stranded RNA. Similar to TLR3, TLR7 and TLR8 are localized on endosomal inner membrane.12 Release of RNA genome into the endosome is required for TLR7 and TLR8 to recognize viral replication, uncrating, or degradation (Fig. 2). As one of the most sensitive anti-viral receptors, both TLR7 and TLR8 recognize almost all RNA viruses, such as dengue virus and influenza virus.1 Interestingly in human system, type I IFN which are produced by plasmacytoid dendritic cells (PDCs) specifically induce TLR7 and MyD88 expression in naive B cells. This induction makes the cells to have more sensitivity against virus invasions.36 Among numerous ssRNAs in the cytoplasm, including mRNAs, viral ssRNAs and other oligonucleotide RNAs, only viral ssRNA and nucleic acid agonists can be recognized by the presence of uridine ribonucleotide and initiate innate immune system via TLR7.37 Similarly, guanosine (G)- and uridine (U)-rich ssRNA oligonucleotides induce signals via TLR8.38 These results, therefore, suggest that the specific region of viral ssRNA is the physiological ligand for TLR7 and TLR8.
TLR9
TLR 9 in the endosomal membrane plays a central role in the host defense against viral infection.39 It is a receptor for sensing unmethylated CpG sequence motif, which is found abundantly in some viral genomes, such as dsDNA virus and HSV.4 In fact, CpG binds to TLR9 via ionic interaction with negatively charged phosphate backbone of DNA.40 In addition, the presence of dinucleotide at 5'-end is critical for receptor recognition more than that at 3'-end.40 When animal cells are infected with dsDNA virus, viral dsDNA is recognized by TLR9 and induces the production of type I IFNs through signal transduction pathway. Interestingly, among the TLRs, only TLR7 and TLR9 are expressed in putative lymphoid DC precursors, although immature myeloid DCs and myeloid DC precursors don't exhibit these patterns in human cells. Therefore, putative lymphoid DC precursors are also called as type I IFNs (alpha/beta/omega)-producing cells (IPCs), because of their IFN-producing ability. This ability may be related with TLR9 expression, allowing the cells to recognize virus infection and induce type I IFNs.39
TYPE I INTERFERONS PRODUCTION VIA TOLL-LIKE RECEPTOR SIGNALING
TLR signaling is mediated by adapter proteins and protein kinases, ultimately leading to the activation of IFN regulatory factor, IRF3, 5 and 7, or NF-κB family.11,16,41 Because these factors have nuclear localization signals (NLSs), they are able to enter the nucleus. As a major transcription factor for anti-viral activity, NF-κB is thought to play an important role in the induction of pro-inflammatory molecules, such as IL-1β, and tumor necrosis factor α (TNF-α), upon cellular responses against the virus infection.42 IRF5 is a strong transcription activator for IFN-α production,41 and IRF7 can induce IFN-α and also IFN-β preferentially.43 Production of type I IFNs is possible because of translocation of these transcription activators into the nucleus, leading to DNA binding at promoters containing interferon-stimulated response element (ISRE). IRF3, -5 or -7 undergoes conformational changes because of phosphorylation (P) at Ser/Thr residues in the carboxyl terminus,44,45 and this leads to C-terminal auto-inhibition and exposes both DNA binding domain (DBD) and interaction domain regions (IAD).45 Because of this conformational change, IRF3 or IRF7 is now capable of forming heterodimer with IRF5. Interestingly, whereas heterodimer of IRF5 and IRF3 still has a function of transcription activator, IRF5/IRF7 heterodimerization does not lead to a functional complex binding to the IFN-α promoter. These results suggest that IRF5 may be a part of the regulatory networks (Fig. 2).45
TYPE I INTERFERON PRODUCTION AND ANTIVIRAL ACTIVITIES
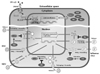
NF-κB activation induces the synthesis of type I IFNs, IL-1β and TNF-α, which exert antiviral and immune stimulatory activities (Fig. 2). Moreover, IRF families-dependent pathway leads to the secretion of type I IFNs such as IFN-α and -β.46 Secreted type I IFNs bind to its receptor, IFNAR, which is composed of subunits IFNAR-1 and IFNAR-2, and make an auto- or paracrine activation.47 Then, signal transduction pathway is operated by phosphorylation of signal transducer and activator of transcription 1 and 2 (STAT1 and STAT2) molecules by Janus tyrosine kinases (JAK).48 Phosphorylated STAT1 and STAT2 form heterodimer, and the latter is translocated into nucleus and then bound to ISRE. Antiviral effectors such as Mx, PKR, RNase L, OAS and ISG are transcriptionally activated to be expressed (Fig. 3).49
Mx GTPase
Mx GTPase, one of the IFN-inducible antiviral effectors, has a strict dependency on strong control of the type I IFNs for their expression.50 In IFN-deficient mice, Mx is also deficient. Similar to the receptor of type I IFNs, IFNAR deficient mice are also deficient for Mx. As previously stated, Mx plays an essential role in host defense against various viruses, especially influenza A virus.51 In support of the above notion, evidence indicates that Mx-neutralizing antibody treatment allows orthomyxoviruses to replicate. Therefore, Mx has a critical antiviral activity against viruses. Mx consists of three domains such as N-terminal GTPase domain, middle linker domain, and C-terminal GTPase effector domain (GED). Each domains are highly conserved among the species. Central interacting domain (CID) and a C-terminal leucine zipper (LZ) can recognize viral components, and has to go through polymerization. The GED which has a critical role in polymer formation has been revealed by experiments with Mx variants52: Mx captures viral nucleocapsid proteins in the cytoplasm and blocks their movement into the nucleus, thereby controlling viral replication by inhibition of nuclear import and further their assembly (Fig. 3).53
RNA-dependent protein kinase
RNA-dependent protein kinase (PKR) is also known as IFN-inducible protein. Active PKR phosphorylates eukaryotic initiation factor 2α (eIFα) and leads to inhibition of viral gene translation, leading to regulated viral protein synthesis.54 In particular, phosphorylation of eIF2α is required for resistance against HSVs55 and rotavirus.56 PKR is activated by binding of viral dsRNA through two N-terminal dsRNA binding motifs (dsRBM), resulting in homodimerization between viral dsRNA and dsRBM.57 As a ligand for PKR, most viral dsRNAs are synthesized in virus-infected cells when viral replication cycle is operated. In RNA viruses, influenza virus for an example, has replicative forms that are intermediate for the synthesis of new genomic RNA copies. In DNA viruses, vaccinia virus, adenovirus or herpes simplex virus for examples, has open reading frames (ORFs) in opposite orientation, so that they are able to produce overlapping mRNA transcripts that can fold to form dsRNA structures, thus offering ligands for PKR.58 IFN-α transgenic mice show an elevated expression level of PKR, and mice lacking a functional PKR pathway are resistant no longer to infection of HSV-2 (Fig. 3).59
Oligo-adenylate synthetase and RNase L
Oligo-adenylate synthetase (OAS) and RNase L are also IFN-inducible antiviral effectors, and they exhibit remarkable antiviral activities against HSV type I when treated with type I IFNs.60 Activity of RNase L is strongly affected by OAS, and activated RNase L leads to viral RNA degradation as intrinsic cellular defenses.61 OAS protein is accumulated in the cytoplasm as an inactive monomer, and dsRNAs containing the motif of NNWWNNNNNNNNNWGN activates OAS.62 Activated OAS converts ATP into 2',5'-linked oligomers of adenosine (2-5A).63 Binding of 2-5A to RNase L triggers dimerization of monomers through their kinase-like domains, thereby enabling RNase L to cleave viral RNAs. It is possible to degrade viral RNA, because RNase L is able to cleave the viral ssRNA at the position of 39 of UpUp or UpAp sequences during the virus infection. On the other hand, RNase L cleaves some self mRNAs and produces small RNAs which act as a ligand for intracellular receptors such as RIG-1 and MDA-5. These receptors also induce type I IFNs and make it possible to acquire antiviral activities. In fact, RNase L-deficient cells are more permissive to viruses such as paramyxovirus, Sendai virus, picorna virus, encephalomyocarditis virus (EMCV), West Nile virus and Coxsackie virus B4 (Fig. 3).64,65
Interferon-stimulated gene
ISG has a role of antiviral activities, and there are several subtypes such as ISG15, ISG17 and ISG20.20,66,67 ISG15 can be conjugated to target proteins through its C-terminal LRLRGG motif which is referred to ISGylation. However, the consequence of ISGylation of target proteins and its cellular functions remain largely unclear.68 Although most of the ISG's activities are not yet clearly elucidated, ISG15 has been reported to inhibit degradation of IRF3 by viral components. IRF3 induces IFN-β gene expression and activates host immune systems,69 and ISG15 deficient mice are susceptible for infection with several viruses, including influenza viruses, Sindbis virus and HSV-1.68 Therefore, it is important to identify the role of ISGylation in the type I IFNs-mediated antiviral response (Fig. 3).
CONCLUSION
Intrinsic cellular defenses arise when PAMPs are recognized by PRRs. Then, innate immune response is rapidly activated to eliminate pathogens and activate adaptive immunities. If there are no activities of innate immune response, no other individuals can be free from invading pathogens. In innate immunity, TLR is an essential component to resist the invasion of pathogens. Especially, TLR2, TLR3, TLR4, TLR7, TLR8 and TLR9 sense viral ligands and give a warning for viral infection by signaling pathway. When viruses infect any cells, attachment entry, uncoating, viral genome replication, viral protein production, their assembly and release are provoked by the following procedure: TLRs detect viral components on each steps of viral infection cycle. It is the reason why TLRs are expressed on the specific location and have different ECDs for each viral ligand. As an outcome of TLR signaling, type I IFNs play antiviral activities through inducing viral replication inhibitory components. IFN-α and IFN-β affect not only sensitized cells, but also neighbor cells, to maintain antiviral states by auto- or paracrine modes. Then, IFN-inducible antiviral effectors such as Mx GTPase, PKR, RNase L, OAS and ISG are produced, and they prevent virus replication. Till now, the signaling pathway of TLR which leads to IFN production has been considered to be important for antiviral activities because of virus to inhibit IFN production. Nevertheless, more studies are needed to enhance our immune systems and eliminate viral pathogens.

 XML Download
XML Download