

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Proteases are now considered not only to be enzymes that conduct hydrolysis of peptide bonds linking amino acids, but also signaling molecules that play an important role in homeostatic regulation in mammals and numerous pathological conditions.1,2 In the skin, three families of proteases have been found in the stratum corneum (SC), including the epidermal specific serine proteases, kallikrein 5 (SC tryptic enzyme, SCTE) and kallikrein 7 (SC chymotryptic enzyme, SCCE), as well as cysteine proteases, including cathepsin C, L, and V (SC thiol protease), and at least one aspartate protease, cathepsin D.3-7 These proteases are tightly regulated by specific protease inhibitors and mediate various cellular responses in the skin, such as inflammation and immune responses, host defense, chemotaxis, cytokine expression, vascular function, tissue repair, and apoptosis.8 In addition to endogenous proteases, exogenous proteases from allergens such as house dust mites, cockroaches, certain bacteria, and fungi can also signal the epidermis. A number of biological activities of proteases are mediated, at least in part, by the activation of its receptors, protease-activated receptors (PARs).9 Recent works have indicated that PAR-2, as a sensor for endogenous as well as exogenous proteases, plays numerous physiological and pathophysiological roles in the skin.8,10 In addition, there is increasing evidence that protease and PAR-2 play an important role in the maintenance of epidermal permeability barrier homeostasis.7,11,12 Moreover, abnormal expression or activity of serine proteases and PAR-2 has been associated with several inflammatory skin disorders involving barrier abnormalities, including atopic dermatitis, netherton syndrome (NS), psoriasis, and peeling skin syndrome.13-18 In this review, we will discuss the role of protease/PAR-2 signaling in epidermal permeability barrier homeostasis, as well as its contribution to the pathogenesis of atopic dermatitis (AD).
SERINE PROTEASES AND THEIR INHIBITORS IN SKIN
Human tissue kallikreins (KLKs) are the largest family of trypsin- or chymotrypsin-like secreted serine proteases.13 Eight KLKs, including KLK5, -6, -7, -8, -10, -11, -13, and -14 are known to be expressed in the epidermis and skin appendages, such as sebaceous gland.19 Among these, only KLK7 exhibits chymotrypsin-like substrate specificity; the other KLKs exhibit trypsin-like serine proteases. It is well known that KLK5 and KLK7 are the major active KLKs in the SC, regulating the desquamation process through corneodesmosomal cleavage and lipid barrier formation by degrading lipid processing enzymes.19-21 In addition, KLK5 and KLK7 have been shown to control the enzymatic processing of cathelicidin, thereby affecting its antimicrobial activity and inflammatory responses.22 KLK14 has also been detected in its active form in the SC and is thought to be a candidate protease involved in the process of desquamation, however, its precise role has not been elucidated.23,24 KLK5 and KLK7 are stored in the form of proenzyme in the lamellar bodies (LBs) along with a substrate of KLK7, corneodesmosin and their inhibitor, lymphoepithelial Kazal-type-related inhibitor (LEKTI).25 Upon release into the stratum granulosum (SG)-SC interface, these pro-KLK zymogens are activated through a KLK proteolytic activation cascade.26 KLK5 can be activated by itself or by KLK14 and then activate several other pro-KLKs; therefore KLK5 is thought to be the key protease for the initiation of the KLK cascade.24 Significantly, KLK5 and KLK14 are known to activate PAR-2, thereby modulating epidermal permeability barrier homeostasis, immune and inflammatory responses, skin pigmentation, itching sensations, as well as tumor surveillance.10,27 KLK8 has been reported to be localized in the LBs with KLK5, KLK7 and their inhibitors and secreted in the SG-SC interface, regulating desquamation and epidermal proliferation.28,29 Recent studies have investigated factors regulating the expression of KLKs in the skin and reported that epidermal calcium ions, vitamin D3, and retinoic acid independently regulate the expression of KLK5 and KLK7 in normal human epidermal keratinocytes.30 An increase in extracellular calcium induced KLK5 and KLK7 expression with induction of differentiation markers, suggesting that the expression of KLKs is induced during epidermal differentiation. In contrast, both 9-cis retinoic acid and 13-cis retinoic acid increased KLK5 and KLK7 expression, independently of keratinocyte differentiation. 1,25 (OH)2 vitamin D3, which is well known to induce cathelicidin expression, was also demonstrated to stimulate the expression of KLK5 and KLK7, which are co-localized with cathelicidin.
In addition to KLKs, the two transmembrane serine protease, matriptase (MT-SP1) and prostasin have been identified in the uppermost epidermal layers and postulated to be involved in epidermal barrier formation and SC hydration via epithelial sodium channel-induced intracellular calcium influx and consequent activation of calcium-dependent proteases and transglutaminases (TGMs).31 Moreover, the observation that mice deleted for matriptase showed impaired desquamation suggests that matriptase is also involved in the process of desquamation.32
A number of serine protease inhibitors are present in the skin, and regulate proteolytic activity in order to prevent excessive serine protease cascade, while also maintaining permeability barrier homeostasis. Among several serine protease inhibitors observed in the SC, such as secretory leucocyte protease inhibitor (SLPI), elafin skin-dericed antileukoproteinase (SKALP), and plasminogen activator inhibitor type 2, which are normally cross-linked to the cornified envelope, LEKTI-1, a secreted serine protease inhibitor, is thought to be the major player in the SC. LEKTI is encoded by SPINK5 (serine protease inhibitor Kazal-type 5) and its multidomains exhibit diverse inhibitory effects toward trypsin, plasmin, subtilisin A, cathepsin G, and human neutrophil elastase.33 In addition, previous in vitro studies demonstrated that LEKTI fragments could inhibit KLK5, -6, -7, -13, and -14.31 LEKTI-1 is stored in specific intracellular LB cargoes, separated from the KLKs, until it is secreted and released into the SC-SG junction to colocalize with KLK5 and KLK7 and inhibit their activity. The interaction strength between LEKTI-1 and KLKs is regulated by the SC pH gradient, with its highest inhibitory capacity at a neutral pH and decreased activity in an acidic pH environment, such as in the superficial SC layer. This implies that a normal SC pH gradient regulates the controlled release of active KLKs in the superficial SC layer and prevents premature desquamation in the deep SC layer, where the pH is neutral.34 These findings support the fact that AD, characterized by altered skin surface pH, and NS, characterized by genetically defective LEKTI, display similar phenotypic features, including premature and excessive desquamation. Recently, LEKTI-2, the product of SPINK9, has been identified to be focally localized at the SG and SC at palmar and plantar sites in close proximity to KLK5.35 It was found that recombinant LEKTI-2 inhibited KLK5 but not KLK7 and -14, suggesting that LEKTI-2 contributes to the regulation of the desquamation process by inhibiting KLK5 proteolytic activity. Other serine protease inhibitors and their roles are summarized in Table 1.36-38
PAR-2 IN SKIN
PAR is a G-protein coupled receptor, characterized by a unique mechanism of self-activation following specific proteolytic cleavage of their extracellular domains.39 Until now, four PAR members have been identified. PAR-1, -3, and -4 are known to be activated by thrombin, and thereby involved in homeostasis and thrombosis, whereas PAR-2 is activated by trypsin-like serine proteases, but not by thrombin.10,40 PAR-2 is known to be widely distributed throughout the mammalian body. In the skin, PAR-2 is abundantly expressed by almost all cell types, especially by keratinocytes. In addition, endothelial cells, fibroblasts, sensory neurons, and inflammatory cells such as mast cells, T lymphocytes, eosinophils, neutrophils, monocytes, macrophages, and dendritic cells are also reported to express functional PAR-2.10,40 Previous studies demonstrated that PAR-2 was expressed in the suprabasal layers of both human and murine epidermis, and that this expression was most prominent in the granular layer, implying that PAR-2 expression might depend on the state of epidermal differentiation.7,40 As opposed to normal skin, the lesional skin of atopic dermatitis has been shown to express high levels of PAR-2 also in the lower epidermal layers.21,40,41 PAR-2 as well as KLK14 has been shown to be widely distributed in lesional skin in rosacea, another inflammatory skin disease.21 Taken together, this evidence suggests that PAR-2 expression may be induced by cutaneous inflammation. In addition, PAR-2 expression has been reported to be regulated by ultraviolet irradiation and involved in melanosome transfer.42
Various endogenous serine proteases including trypsin, mast cell derived tryptase, KLK5, -6, and, -14, matriptase-1 [membrane-type serine protease-1 (MT-SP1)], human airway trypsin-like protease (HAT), cathepsin G, and factor Xa have been demonstrated to activate PAR-2. In addition, some kinds of pathogenic organisms with proteolytic activity such as house dust mites, cockroaches, pollens, molds or bacteria may also be exogenous activators of PAR-2. Upon activation, PAR-2, which is mainly localized to lipid raft domains under basal status, is known to be endocytosed and degraded.7,43 As a G-protein coupled receptor, PAR-2 is known to have a common signaling pathway, including the activation of phospholipase C, which results in the formation of ionsitol triphosphate and diacylglycerol, followed by calcium mobilization.39 In vitro, PAR-2 activation by PAR-2 agonist peptide has been demonstrated to provoke transient intracellular calcium mobilization in primary keratinocytes, suggesting that PAR-2 could regulate the proliferation and differentiation of keratinocytes.44,45 The precise role of PAR-2 in epidermal barrier homeostasis is discussed below.
PROTEASE/PAR-2 AND EPIDERMAL BARRIER
KLKs and their inhibitors are co-localized in the LBs and secreted to the SG-SC junction. The enzyme activity of various proteases in the skin and the inhibitory activity of protease inhibitors are regulated by the SC pH gradient. These findings imply that proteases play an important role in epidermal permeability barrier homeostasis, and recent studies have further shown the importance of proteases and PAR-2 signaling in the barrier function of the skin. Proteases exert various cellular responses, some of which may be, in part, mediated via activation of PAR-2.
Non-PAR-2-mediated function
Filaggrin processing
Filaggrin aggregates keratin intermediate filaments to form the cornified cell envelope, thereby providing the structural support and mechanically resilient skin barrier. In addition, these polypeptides are degraded into natural moisturizing factors (NMF), and contribute to water retention within the SC layers, helping to maintain skin hydration.46,47 A number of proteases, including PEP-1, µ-calpain, furin, prostatin, matriptase, and caspase-14, elastase2 (ELA2), have been demonstrated to be involved in the proteolytic processing of filaggrin and profilaggrin, which leads to epidermal differentiation, barrier formation and hydration.32,48-52 Recently, the two membrane-bound serine proteases matriptase and prostasin have been reported to be involved in the processing of profilaggrin by activating the epithelial sodium channel (ENaC) which causes a calcium influx through a voltage-gated calcium channel, thereby inducing the activation of calcium-dependent proteases.31 Matriptase, or MT-SP1 is a type II trans-membrane serine protease and is now considered to be an essential component of the profilaggrin-processing pathway, in accordance with the observation that neonate Matriptase/MT-SP1-deficient skin displayed a loss of mature filaggrin monomer.32 Matriptase is also known to be co-localized with another membrane-bound serine protease, prostatin (CAP1/PRSS8), and could activate prostatin in vitro, resulting in the initiation of a proteolytic cascade toward ENaC activation.53
Desquamation
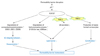
Desquamation involves the enzymatic process of degradation of corneodesmosomal proteins including corneodesmosin (CDSN), desmoglein 1 (DSG1), and desmocollin 1 (DSC1). It is well known that KLKs, especially KLK5 and KLK7, act as main players in desquamation through a pH-dependent protease signaling cascade (Fig. 1).20 KLK7 directly cleaves CDSN and DSC1 but is unable to degrade DSG1, while KLK5 can.54 During KLK proteolytic activation cascade, pro-KLK5 is activated by KLK14 and by KLK5 itself, and the active KLK5 then activates pro-KLK7 and pro-KLK14; thus, KLK5 is believed to be the cascade initiator.24 Besides KLK5, -7, and -14, it has been reported that KLK8 is also involved in skin desquamation through a protease cascade reaction leading to the degradation of DSG1 and CDSN.29 Recently, KLK6, -13, and -14 have been found to degrade DSG1 and be inhibited by LEKTI, suggesting that these KLKs are also potential desquamatory enzymes.54 The desquamation process is tightly controlled by the epidermal pH gradient. Skin pH regulates not only the activity of KLK but also the binding of KLK to LEKTI fragments.55 At a neutral pH of the SG-SC junction, KLK is tightly bound to LEKTI fragments, however, with a decrease in pH (acidic pH of the SC) the dissociation of KLK from LEKTI becomes more frequent, releasing free KLKs into the outer layer of the SC.55 However, at the acidic pH of the superficial SC layer, KLKs exhibit lower activity than at a neutral pH. This bidirectional regulation of KLKs and their inhibitors by pH is important to maintain proper skin desquamation.13 In addition to serine proteases, cystein protease, cathepsin V, and cystatin M/E, an inhibitor of asparaginyl endopeptidase legumain (LGMN) and cysteine proteases also controls desquamation.56,57 Cathepsin V has been known to degrade DSG1, DSC1, and CDSN with a higher proteolytic activity at an acidic pH, suggesting that cathepsin V is a major player in desquamation under basal conditions with a normal SC acidic pH.56 Cystatin M/E regulates desquamation by inhibition of cathepsin V as well as LGMN, which regulates pH-dependent processing of (pro)-cathepsins. In addition, cystatin M/E regulates crosslinking of structural proteins by transglutaminase (TGM) 3 during epidermal differentiation by controlling cathepsin L and LGMN activities.56 A recent study has shown that the cystatin M/E and cathepsin V were expressed to a lesser degree in lesional skin of AD, suggesting that disturbance of the cystatin M/E-cathepsin pathway could contribute to abnormal skin barrier function in AD.58
Degradation of lipid processing enzyme
The permeability barrier function of the SC is provided by lipid bilayers and corneocytes. The lamellate structure of SC intercellular lipids is formed by the delivery of lipid precursors to the SG-SC junction by LBs and the proper processing of these precursors by their extracellular processing enzymes. It has been demonstrated that serine proteases have a central role in the formation and maintenance of the epidermal lipid barrier by degrading the key lipid processing enzymes required for normal permeability barrier homeostasis and PAR-2-mediated manipulating LB secretion (Fig. 1).7,21 Hachem, et al.21 reported that increased serine protease activity provoked by sustained SC neutralization with superbase application in murine skin leads to degradation of both β-glucocerebrosidase (β-GlcCer'ase) and acid sphingomyelinase (aSMase) and consequent defect of epidermal barrier function, which was reversed by coapplied SP inhibitors. In addition to this direct proteolytic effect, serine proteases could affect LB secretion via PAR-2 activation.7
Control of antimicrobial function in skin
Antimicrobial peptides (AMPs) are important molecules that comprise the innate immune defense system of the skin. Besides their antimicrobial activity, AMPs in skin have been known to have multiple functions, including modulation of host imflammatory responses and promotion of wound healing.59 Moreover, previous study has suggested that AMPs are associated with permeability barrier function by showing a significant delay in permeability barrier recovery after tape stripping and structural abnormalities in LB contents in cathelin-related antimicrobial peptide (CRAMP), the murine homologue of LL-37, knockout mice.60 Recently, proteases have been demonstrated to regulate the antimicrobial activity or inflammatory effect of AMPs through proteolytic degradation of these peptides. KLK5 and KLK7 were shown to be co-localized with cathelicidin and to control enzymatic activation of the cathelicidin precursor (hCAP18) and also to influence processing into shorter peptides with alternate biological activity.22 The observation that the epidermal extracts of SPINK5-deficient mice show increased antimicrobial activity as compared with the controls indicates that the processing of cathelicidin by highly active serine proteases in SPINK5-deficient mice skin augmented antimicrobial activity.22 Recent study has demonstrated that the expression of KLK5 and cathelicidin was up-regulated in the lesional skin of patients with rosacea with altered abundance and the processing of cathelicidin peptides compared to normal individuals.61 It was also suggested that the high levels of abnormally processed cathelicidins observed in rosacea patients are a result of a post-translational processing associated with increased serine protease activity and that these proteolytically processed forms of cathelicidin peptides trigger skin inflammation in rocasea.61
PAR-2- mediated function
Permeability barrier homeostasis
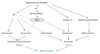
The involvement of PAR-2 in epidermal permeability barrier homeostasis by mediating signaling from serine proteases in the skin has been recently evidenced.7,11,12 Although controversial, recent studies have shown that PAR-2 localized in the suprabasal layers of mouse and human epidermis with high levels of PAR-2 in the stratum granulosum, suggesting that PAR-2 might act as the primary sensor of barrier-initiated serine protease activity.7,62,63 Acute barrier disruption leads to an increase in SC pH from basal levels of 5.0-5.5 toward a more neutral pH, which, in turn, increases the activity of serine protease, which has an optimal activity at neutral pH.11,64 This increase in serine protease activity activates PAR-2 on keratinocytes of SG, resulting in decreased LB secretion and a consequent decrease in the formation of caveolae and lipid raft (Fig. 2).7 It was reported that topical PAR-2 agonist peptide significantly delayed barrier recovery and inhibited LB secretion following acute barrier disruption in murine skin, whereas PAR-2 knockout mice showed increased LB secretion and accelerated barrier recovery following acute disruption as compared to wild-type littermates.7 In addition, inhibition of PAR-2 activation, by topical application of either serine protease inhibitor or PAR-2 specific antagonist significantly accelerated the barrier recovery rate after acute barrier disruption.65 These results implicates that PAR-2 activation might be an important signal for regulating LB secretion during the repair response after barrier disruption. While serine protease/PAR-2 signaling negatively affects permeability barrier homeostasis by inhibiting the restoration of lipid barrier (mortar), it could also act as a positive regulator in the permeability barrier recovery by accelerating cornification, which then induces the formation of corneocytes (bricks) (Fig. 2).11 Recent studies have demonstrated that proteolytically active allergens, house dust mites and cockroaches, also activated PAR-2 and delayed barrier recovery via PAR-2 signal-mediated inhibition of LB secretion.12 By showing the abnormal distribution of calcium ions after barrier disruption in skin where allergens had been applied, the authors suggest that PAR-2 signaling-mediated modulation of calcium ions in the skin could be one of the mechanisms involved in the regulation of LB secretion by PAR-2.12
Inflammation
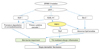
The role of PAR-2 in the regulation of inflammation has been widely investigated. During cutaneous inflammation, potential endogenous activators of PAR-2, including leukocyte elastase, mast cell tryptase, proteinases of the trypsin-family produced by keratinocytes (trypsinogen-4), and proteinases of the fibrinolysis cascade such as factor VII/Xa, are released; these then activate PAR-2 on keratinocytes, endothelial cells, inflammatory cells, and dermal sensory nerves to amplify the inflammation via the up-regulation of inflammatory mediators.66 It has been demonstrated that PAR-2 agonist peptide induces intercellular adhesion molecule-1 (ICAM-1) expression in primary human keratinocytes via the activation of nuclear factor-kappa B.67 Another study reported that stimulation of PAR-2 on keratinocytes increased the secretion of interleukin (IL)-6 and granulocyte-macrophage colony-stimulating factor (GM-CSF).68 In keratinocytes, PAR-2 activation also leads to a secretion of IL-8/CXCL8, promoting granulocyte and T-cell recruitment.69 The observation that PAR-2 deficient mice show diminished ear swelling and inflammatory infiltrates in a model of contact hypersensitivity indicates that PAR-2 mediates inflammation in allergic dermatitis.70 In addition, PAR-2 has been observed at increased levels in lesional skin of patients with AD, suggesting that PAR-2 plays a role in inflammatory dermatosis.15,17 In addition to these proinflammatory responses, PAR-2 also has been shown to be involved in the T-helper type 2 (Th2) mediated allergic inflammation. Thymic stromal lymphopoietin (TSLP), a Th2-associated cytokine produced by inflammatory cells as well as epithelial cells, is known to induce Th2 cell recruitment and allergic inflammation via dendritic cell stimulation in response to allergen challenge, microbial infections, and inflammation.71 TSLP transgenic mouse has been shown to develop an AD-like skin disease, which suggests an important role of TSLP in initiating and perpetuating Th2 immune responses in AD.72,73 A recent study reported that SPINK5 knockout mice expressed TSLP in the epidermis at a higher rate than that of wild-type epidermis, and demonstrated that KLK5 directly activates PAR-2, which in turn induces nuclear factor κB-mediated over-expression of TSLP. This implies that LEKTI deficiency-induced serine protease/PAR-2 activation triggers proinflammatory as well as proallergic inflammatory responses in the development of AD-like phenotype of NS (Fig. 3).74
Pruritus
Skin nerve fibers are known to have functional PAR-2, and it has been proposed that PAR-2 plays an important role in pruritus. During neurogenic inflammation, various endogenous serine proteases such as tryptase from mast cells and trypsins from keratinocytes activate PAR-2 on sensory nerve ending to release calcitonin gene-related peptide (CGRP) and substance P (SP). These neuropeptides induce vasodilation, edema, and leukocyte recruitment, resulting in neurogenic inflammation in the local skin lesions.75 PAR-2 is not only expressed in the peripheral nervous system but also in the central nervous system, spinal cord and brain. Therefore, PAR-2 signaling also stimulates the release of neuropeptides from central nerve endings at the spinal cord level, thereby activating CGRP receptor and SP receptor (NK1R) to transmit itch responses to the central nervous system.76 In addition, skin exposure to exogenous microbial proteases could also induce itch and inflammation via PAR-2. Recent study reported that the mice that over-expressed epidermal KLK7 displayed massive itchy behavior.77 Another study demonstrated that trypsin-induced scratching behavior in mice was inhibited by a PAR-2 blocking peptide, suggesting the role of serine protease/PAR-2 signaling in pruritus.78 Moreover, PAR-2 activation is likely to be involved in pruritus of AD.79 PAR-2 and trypsin have been observed to be expressed at high rates in the lesional skin of patients with atopic dermatitis, and PAR-2 agonist peptides induce pruritus in AD patients.41 In addition, the fact that KLK5, -7, -8, and -14 are present in sweat, (sweat being the most common itch-triggering factor in AD) point to the important role of PAR-2 in pruritus of AD patients.7,80 PAR-2 is reported to interact synergistically with transient receptor potential (TRP) vanilloid-type 1 (TRPV1), which belongs to the superfamily of TRP channels, thereby amplifying itch sensation.81 These findings suggest that serine protease inhibitors or PAR-2 antagonists might be a promising therapeutic tool for the management of itching and help break the vicious itch-scratch cycle in AD.41,75
ABERRANT PROTEASE/PAR-2 SIGNALING IN THE PATHOGENESIS OF AD
Genetic abnormalities in the genes encoding protease/protease inhibitor
There is increasing evidence that genes related to protease/protease inhibitor become deregulated in patients with AD, shifting the balance between proteases and protease inhibitors toward increased protease activity (Fig. 4). It has been demonstrated that there is an association between AD and a four base pair (AACC) insertion in the 3'-untranslated region of KLK7 gene, which increases the half-life of KLK7 mRNA and enzymatic activity of KLK7.82 The rare AACCAACC variant of KLK7 gene showed a more significant association with AD as compared to the common AACC variant. The genetic variant of KLK7 gene was found to be more significantly relevant in patients who did not have an elevated IgE level. The enzyme KLK7 plays an important role in desquamation by cleaving corneodesmosomal proteins. In addition, it was reported that transgenic mice over-expressing KLK7 presented cutaneous manifestations similar to chronic AD.83 This implies that the gain-of-function mutation polymorphism in the KLK7 gene causes a premature breakdown of corneodesmosomes, leading to excessive desquamation and impairment of the epidermal barrier in the development of AD. In addition to abnormalities in the protease-encoding genes, several genomic defects in genes encoding members of the protease inhibitors have been identified in patients with AD. LEKTI is an inhibitor of multiple serine proteases in skin and tightly regulates the enzymatic activities of serine proteases including KLK5, -6, -7, -13, and -14, thereby controlling epidermal barrier function.31
A premature stop codon mutation in the SPINK5 gene, which encodes LEKTI, is known to be associated with NS, a rare ichthyosiform dermatosis characterized by congenital ichthyosiform erythroderma, severe atopic manifesations, and hair-shaft abnormality.84 The SPINK5 gene consists of 33 exons, encoding 15 LEKTI inhibitory domains with selective/specific inhibitory function.85 Previous study attempting to correlate genotype with phenotype in Japanese NS patients has demonstrated that the clinical severity of NS correlates with the residual expression of LEKTI-1.86 SPINK5-deficient mice have been reported to show increased proteolytic activities of KLK5 and KLK7 in the epidermis, abnormal degradation of DSG1, and resultant abnormal corneodesmosome cleavage and premature desquamation, suggesting that LEKTI is a key regulator of KLK5 and KLK7 activity and that defective SC adhesion by epidermal protease hyperactivity is the primary pathogenic event in NS.87 Recently, ELA2, a novel epidermal protease, has been identified in human and mouse skin and conceived as a potential trigger in NS pathogenesis by misprocessing of filaggrin and lipid.88 Being localized to the keratohyalin granule, ELA2 directly degrades (pro-) filaggrin and disrupts the lipid lamellae formation. LEKTI was demonstrated to inhibit ELA2 activity indirectly, by controlling KLK5-mediated cleavage of pro-ELA2, and indeed, ELA2 was observed to be hyperactive in the LEKTI-deficient epidermis of NS patients and SPINK5-deficient mice.88 Transgenic mice over-expressing ELA2 also mimic the clinical features of NS patients with transient ichthyosiform dermatitis and impaired lipid barrier, implying that ELA2 is a critical epidermal protease that regulates epidermal barrier homeostasis, involving the pathogenesis of NS. Several studies reported that the polymorphisms in the SPINK5 gene are also associated with AD.84,89 Glu420Lys single-nucleotide polymorphism in the SPINK5 gene was observed to be associated with AD and six other SPINK5 gene polymorphisms were also found in Japanese patients with AD.84,89 In addition, another protease inhibitor, cystatin A-encoding gene (CSTA gene) polymorphism (+344c variant) has been reported to be associated with AD.90 Cystatin A is a cysteine protease inhibitor and is known to inhibit the endogenous cathepsins B, -H, and -L and the exogenous proteases from house dust mites, such as dermatophagoides pteronyssinus (Der p) 1 and dermatophagoides farinae (Der f) 1. This +344c variant in CSTA gene results in the reduced levels of cystatin-A in the skin surface and sweat, allowing exogenous proteases to break down the integrity of the SC, which in turn enhances the penetration of allergens, triggering an aggravation of AD. These genetic variants in protease and protease inhibitor genes can be combined in some patients with AD, resulting in a more severe defect in the skin barrier.
Elevated pH
The skin has a distinct pH gradient across the SC, with a progressively more acidic pH from the deeper layer to the outer layer of the SC.91 Studies have demonstrated the 'acid mantle' within the SC plays a critical role in regulating epidermal permeability barrier homeostasis, the epidermal antimicrobial barrier, and SC integrity/cohesion by regulating the activity of various enzymes including serine proteases and lipid-processing enzymes.91 Skin pH is known to be generated by the endogenous processes, including transurocanic acid production from histidine, free fatty acid production via secretory phospholipase A (sPLA2), and the sodium-proton antiporter-1 (NHE-1)-mediated H+ secretion.92 Numerous endogenous factors, such as moisture, sweat, sebum, anatomic site, genetic predisposition and age as well as environmental factors, such as soap/detergents, topical irritant, antibiotics, and cosmetic products, and occlusive dressings can affect skin pH.93 In the lesions of patients with AD, the skin pH has been reported to be significantly elevated.94 The genetically determined barrier deficiency and various environmental factors are thought to be associated with the increased skin surface pH in AD lesions.
Since Palmer, et al.95 first identified the loss-of-function mutation in the gene encoding filaggrin (FLG) as a strong predisposing factor for AD, a number of additional studies have reported other mutations of FLG genes, including 20 mutations in European populations and 17 in Asian populations.96 From these reports, it is now conceived that mutations of the FLG gene are the most significant genetic factors in AD and the resultant genetically determined epidermal barrier defect is the primary event triggering immunologic pathogenesis in AD. Filaggrin itself contributes to the epidermal barrier integrity by cross-linking the keratin filaments and the being degraded into a combined pool of highly hygroscopic amino acids, called the NMF, thereby contributing to the SC hydration. It is also postulated that filaggrin contributes to the formation of acid mantle within SC through the generation of urocanic acid via filaggrin-histidine-urocanic acid cascade.97 Therefore, filaggrin deficiency in AD lesion leads to defects in the formation of cornified envelope and a decreased ability of maintaining SC hydration and a concomitant elevation of pH. The elevated pH enhances the activity of KLK5 and KLK7, which have optimal activity at neutral pH, resulting in the over-degradation of corneodesmosomes and a decrease in SC integrity and cohesion. In addition, the abnormally neutral skin pH inhibits the activity of lipid-processing enzymes, including β-GlcCer'ase, aSMase, and sPLA2, which are essential in the production of ceramide and free fatty acids and causes impaired lipid processing and defects in the lipid barrier. SC acidity is also important in epidermal antimicrobial barrier function, inhibiting the growth of pathogens. Growth of Staphylococcus aureus, which colonizes the lesional skin of AD, is normally inhibited at low skin pH; therefore the elevated pH in AD lesional skin leads to bacterial growth, resulting in allergic inflammation and aggravation of AD.94 In addition to these genetic factors, various environmental factors can increase skin surface pH in AD patients. For instance, the frequent use of soaps/detergents seems not only to aggravate AD through directly irritating the skin by removing skin surface lipids, but also raises skin pH because most detergents are alkaline; but even at neutral, overuse of soap and detergent can have a negative effect on epidermal barrier function.
Exogenous proteases
Various contact allergens and aeroallergens are considered important factors in the initiation and aggravation of AD. Proteolytic activity from allergens has been known to play a role in the pathogenesis of allergic diseases including allergic rhinitis, asthma, and AD through inducing Th2 allergic inflammation and directly affecting the structure and function of epidermal barrier, thereby facilitating further penetration of allergens through the defective skin barrier.98 House dust mite and cockroach allergens, the most important environmental factors in the pathogenesis of AD, have been shown to have proteolytic activity. House dust mite allergens have been reported to contain several cysteine and serine proteases. Serine proteases from mite allergens, Der p 3 and Der p 9, are known to activate PAR-2 on keratinocytes to produce cytokines and contribute to the pathogenesis of AD, whereas Der p 1 with cystein protease activity stimulates inflammation via a PAR-2-independent mechanism.99 The cockroach allergens, another type of major aeroallergen, are also reported to activate PAR-2 on keratinocytes. Jeong, et al.12 reported that the topical application of house dust mites and cockroach allergens to barrier-disrupted skin delayed barrier recovery and lamellar body secretion in murine and human skin via PAR-2 activation. The topical application of cockroach allergens also showed an inhibitory effect on the epidermal calcium gradient change after barrier disruption, suggesting that the negative effects of cockroach allergens on barrier recovery may be due to the PAR-2 signal-induced epidermal calcium modulation. Recently, Staphylococcus aureus has also been reported to produce extracellular V8 protease, which exhibits a similar specificity of glutamate-specific cleavage and a similar sequence to exfoliative toxins and cause epidermal permeability barrier dysfunction in the skin of nude mice by directly degrading DSG1.100 These imply that proteolytically active allergens could break down the skin barrier via PAR-2-mediated inhibition of lamellar body secretion or PAR-2-non-mediated mechanisms, including degradation of corneodesmosomal proteins and lipid processing enzymes, triggering further allergen penetration through the disrupted epithelial barrier to aggravate Th2-mediated inflammation and possibly switch the non-IgE-associated form of AD (atopiform AD) to the typical IgE-associated form of AD. Recently, several reports have suggested that proteolytically active allergens perturb not only the SC permeability barrier but also the tight junction (TJ), another functional barrier of the skin.101-103 The exfoliative toxin-negative Staphylococcus strains were shown to decrease the expression of TJ and atypical protein kinase C, a key player in TJ assembly with decreased transepithelial resistance in the human keratinocyte cell line.101 Previous studies have reported that house dust mite allergen Der p 1 disrupts intercellular TJ via proteolytic cleavage of TJ adhesion protein, claudin-1 and occludin in airway epithelial cells.102 Pollens, which are known to be associated with allergic rhinitis and conjunctivitis, also contain proteolytic enzymes on their surface and degrade the TJ proteins of human airway epithelial cells.103 These findings suggest that proteases derived from house dust mite, cockroaches, Staphylococcus aureus, and pollens contribute to the pathogenesis of AD by propagating the vicious cycle of protease-mediated permeability barrier defect and increased allergen penetration (Fig. 4).
THERAPEUTIC APPLICATION OF PAR-2 ANTAGONIZING TREATMENT
Identification of the important role of PAR-2 in various AD symptoms, including inflammation, pruritus and skin barrier impairment, has suggested that PAR-2 antagonizing treatment may be a potential therapeutic strategy for AD. In addition, the crucial role of mast cells and mast cell-expressed PAR-2 in various inflammatory diseases also suggests that PAR-2 antagonizing treatment could be applied to several chronic inflammatory diseases, such as asthma,104 rheumatoid arthritis105 and inflammatory bowel diseases (IBD).106 Theoretically, several approaches could be used to antagonize PAR-2 activation, including down-regulation of protease activity, inhibition of PAR-2 expression using siRNA technology, PAR-2 monoclonal antibody or PAR-2 specific antagonist; however, at this time, a practical application of PAR-2 antagonizing treatment has not yet reported.
Down-regulation of protease activity to control PAR-2 activation in skin can be achieved by either topical protease inhibitors or pH controlling agents. We recently reported that the topical application of serine protease inhibitor (soybean tryptic inhibitor) significantly reduced the pruritus symptoms in end-stage renal dysfunction patients, where increased PAR-2 expression in skin was observed. Improvement of skin barrier functions was also observed after protease inhibitor application.107 While this is a relatively easier way to control PAR-2 activation, proteases are responsible not only for PAR-2 activation, but also for other normal homeostatic processes such as desquamation. As a result, use of protease inhibitors may have adverse effects on the skin, which is a major drawback for practical application of protease inhibitors for topical use.
Normal skin surface pH ranges from 5.0 to 5.5, which is relatively acidic compared to the normal physiologic pH, and it is well known that pH gradually increases across the SC. This "acid mantle" is very important for the maintaining skin's homeostasis, including regulating protease activity. In AD, increase of skin surface pH is well recognized and increased proteolytic activity is also observed. In addition to the PAR-2 activation, increased pH is also considered responsible for increased susceptibility to secondary infections in AD patients. As a result, controlling skin surface pH within normal acidic range is a very important therapeutic regimen for AD patients and use of soap or alkaline detergents is strongly discouraged.
The inhibitory activity of the PAR-2 antibody on itching behavior in chronic dry skin itch in an animal model has also been recently reported. Intradermal injection of PAR-2 antibody significantly reduced spontaneous scratching behavior, which alludes the important role of PAR-2 in itching sensation. Further evidence was provided by the increased scratching behavior after PAR-2 activator injection.108 However, a therapeutic antibody against PAR-2 has not yet been reported, and practical application of PAR-2 antibody needs to be further developed.
Among the several approaches for PAR-2 antagonizing treatment, use of small molecule PAR-2 specific antagonist seems to be the most promising method. However, while several PAR-1 specific antagonists have already been reported, the lack of a specific, potent PAR-2 antagonist has hampered more detailed investigation of the role of PAR-2 in various diseases and its practical application. Recently, however, a few PAR-2 specific antagonists and their in vivo and in vitro effects have been reported. ENMD-1,068 (N1-3-methylbutyryl-N4-6-aminohexanoyl-piperazine) was the first reported PAR-2 specific antagonist, and its anti-inflammatory activity was evaluated using both in vivo and ex vivo models. In a carrageenan/kaolin (C/K)-induced arthritis animal model, intraperitoneal injection of ENMD-1,068 showed significant inhibition of joint swelling109 and in ex vivo studies using rheumatoid arthritis synovium and cultured synovial fibroblasts, ENMD-1,068 also showed inhibitory effects on proinflammatory cytokine production in a dose-dependent manner.110 In addition, we have shown that topical application of ENMD-1,068 on barrier disrupted skin significantly accelerated barrier recovery rate, which also suggest the potential application of ENMD-1,068 as a topical agent.12 However, its low potency ranging low millimolar concentration has made it impractical for therapeutic application.
More recently, Kanke, et al.111 have reported several peptidomimetic compounds as having PAR-2 antagonistic activities. Among the compounds, it was reported that K-14585 {N-[1-(2, 6-dichlorophenyl)methyl]-3-(1-pyrrolidinylmethyl)-1H-indol-5-yl)aminocarbonyl}-glycine-Llysinyl-Lphenylalanyl-N-benzhydrylamide) showed the most potent antagonist activity against PAR-2 activating peptide induced cellular responses. Ex vivo responses, such as rat-isolated aorta relaxation, and in vivo responses, such as plasma extravasation in the dorsal skin of guinea pigs and saliva secretion in anaesthetized mice, were also inhibited by a systemic injection of K-14585. Interestingly, it was observed that while K-14585 significantly inhibited the activating peptide-induced PAR-2 activation signaling, serine protease (trypsin)-induced signaling was affected to a much lesser extent. It was suggested that there may be some structural differences between the activating peptide binding site and tethered ligand binding site; this distinction need a further investigation. More interestingly, in a study of K-14585-induced cellular signaling, higher concentrations of K-14585 showed agonistic activity, which still needs further investigation.112 Given these results, practical therapeutic application of K-14585 is currently not achievable.
Currently, the authors are investigating a new PAR-2 antagonist. In a series of in vitro and in vivo studies, the new PAR-2 antagonist, NPS-1577, has shown potential anti-inflammatory activity as well as skin barrier improving activity and anti-hyperproliferative activity on the epidermis (paper in preparation). While detailed investigations related to the action mechanism are currently being performed, NPS-1577 could potentially be used as the first practical PAR-2 antagonist for therapeutic use in the near future.


 XML Download
XML Download