

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Hemophilia A is a hereditary X-linked disorder caused by a deficiency in coagulation factor VIII (FVIII) or functional defect of the gene, resulting in lifelong bleeding disorder. Hemophilia A is observed at a frequency of 1-2 in 10,000 males and characterized by bleeding into soft tissues, muscles and weight-bearing joints.1 The clinical severity of hemophilia and plasma FVIII level are closely related with mild (more than 5%), moderate (1-5%) or severe (less than 1%) phenotypes, and symptomatic patients usually have less than 5% of FVIII levels.2 The current standard therapy is intravenous injection of plasma-derived FVIII concentrates or recombinant FVIII protein. Maintenance of circulating FVIII levels higher than 5% of the normal FVIII concentration may significantly improve the clinical symptoms.3 Even though the replacement therapy has been standard treatment for hemophilia, high cost, unpredictable shortages of recombinant FVIII products and risk of blood-borne virus transmission have been major disadvantages.4 Another problem of replacement treatment is the development of the FVIII neutralizing antibodies in 20-30% of hemophilia patients, rendering further substitution ineffective.5 Therefore, gene therapy is being explored as the next generation therapy for hemophilia patients. Gene therapy is considered suitable for hemophilia treatment because of several reasons. First, hemophilia gene therapy can easily correct this abnormality. Second, wide therapeutic window with low FVIII concentration in 2-5% of normal level will be sufficient to correct the severe phenotype. Furthermore, the site-specific expression is not required to achieve the therapeutic goals.6-8
The development of gene therapy for hemophilia A has been explored by using ex vivo and in vivo gene delivery strategies. Ex vivo gene therapy has the advantage of avoiding the systemic administration of viral vectors. Although therapeutic levels of FVIII could be detected in plasma, the limited survival of implanted cells can lead to gradual decline of gene expression and surgical procedure on ex vivo protocol could be undesirable in hemophilia patients. In vivo gene therapy provides cost-effective treatment compapred to most of the ex vivo protocols. In general, viral vector mediated gene transfer is more efficient than non-viral gene transfer.9 Currently, the vectors used for in vivo FVIII gene transfer include adenoviral vectors, adeno-associated viral vectors, retroviral vectors and lentiviral vectors. Each vector has its own advantages and drawbacks. Lentiviral-based vectors present several attractive features for FVIII gene therapy. The vector transduces the interesting genes into the proliferating and non-proliferating cells at similar efficiencies and mediates stable integration, resulting in sustained expression.10-13 In this study, we evaluated the possibility of hFVIII increase by transduction of lentiviral vectors (LV) into skeletal muscle cells.
MATERIALS AND METHODS
Lentiviral vectors plasmids
The vector plasmids were all derivatives of the pRRL-RRE-cPPT-EF1α-X-PRE-SIN. The transfer plasmid pRRL-RRE-cPPT-EF1α-hFVIII-PRE-SIN containing the human B-domain deleted coagulation FVIII cDNA, driven by EF1α promoter, and pRRL-RRE-cPPT-PGK-LacZ-PRE-SIN containing nuclear localized lacZ, driven by the PGK promoter, were cloned using standard techniques. The packaging construct, pCMVΔR8.74, and the envelope plasmid, pMD.G, have been previously described.14 The rev-expressing plasmid, pRSV-REV, and the packaging plasmid, pCMV.gag.pol.RRE.bpA, were provided by Dr. Frank Park (Medical College of Wisconsin, Milwaukee, WI, USA).
Lentiviral production and assays
The vesicular stomatitis virus (VSV)-G pseudotyped lentiviral vectors were generated as previously described in our lab.15 In brief, transient calcium phosphate transfection of 293T cells was performed using the following amounts of DNA: 10 µg transfer plasmid, 6.5 µg packaging plasmid, 5 µg rev-expressing plasmid and 3.5 µg envelope plasmid. Chloroquine (25 µM) was added to the media prior to transfection. Media were replaced 12 hours after transfection, harvested after further 36 hours of incubations, and then filtered and concentrated by ultracentrifugation. The concentrated viral pellet was resuspended in PBS containing 10 µg/mL polybrene. For the titer of the LV stocks with lacZ gene, serial dilution of concentrated virus was used to infect 5×105 Hela cells in a 6-well plate in the presence of polybrene (8 µg/mL). For the LV containing the human B-domain depleted FVIII gene, the titer of the vector preparation was determined by enzyme-linked immunosorbent assay (ELISA) for the p24 Gag antigen concentration (Alliance; Dupont-NEN).
In vivo gene-transfer protocols
Five weeks old Sprague-Dawley male rats were used in the experiments. Lentiviral vectors containing Lac Z gene as a control or FVIII gene at a dose of 1.5×107 infectious unit (15 ug of p24 Gag antigen) were intramuscularly injected with 10 µg/mL of polybrene into the thigh muscle.
X-gal staining of tissue sections
Four weeks after virus injection, the injected skeletal muscle, liver, spleen, and lungs were harvested and snap frozen using O.C.T embedding medium on dry ice. Sections were made at 10 um thickness, fixed in 0.1% glutaraldehyde, washed in PBS, and subsequently stained in 5-bromo-4-chloro-3-indolyl-β-D-galactoside (X-gal, Invitrogen, Carlsbad, CA, USA) solution overnight. Then, the sections were rinsed in PBS, mounted, studied under a microscope, and photographed.
Measurement of plasma human FVIII concentration
Blood sample was obtained by tail vein catheterization at post-injection 0, 1st, 2nd, 3rd, 4th week and then every 2 weeks up to 12 weeks. Blood samples were transferred into the Eppendorf tubes containing 20% sodium citrate. Plasma was isolated by centrifugation and stored at -80℃ for determination of FVIII activity. The plasma concentration of human FVIII was measured by ELISA as described by the manufacturer (Affinity Biologicals, Hamilton, ON, USA).
RESULTS
In vitro production of human coagulation FVIII in lentiviral vector-transduced Hela cells
To determine the functionality of lentiviral vectors to express hFVIII, Hela cells were transduced by lentiviral vectors carrying hFVIII cDNA, incubated for 3 days and conditioned media was harvested to measure hFVIII concentrations. As shown in Fig. 1, hFVIII concentrations were dose-dependently elevated, following increasing amount of administered virus (n = 2, independent experiments).
β-galactosidase assays
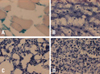
To assess the transduction efficiency of lentivirus in the skeletal muscle, lentiviral vector (LV) carrying bacterial LacZ (dose of 1.5×107 infectious unit) was injected into the thigh muscle. Four weeks after injection, the LV-injected skeletal muscle, liver, lung, and spleen were harvested and sectioned for X-gal staining. Fig. 2 shows strong, localized X-gal-stained nuclei only in the skeletal muscle (Fig. 2A). No X-gal-stained nuclei were found in any other organs, including the liver (Fig. 2B), lung (Fig. 2C), or spleen (Fig. 2D). These results demonstrate that skeletal muscle could be effective target tissue for long-term foreign gene expression.
Plasma FVIII concentrations following LV administration into the skeletal muscle
After virus injection, the plasma FVIII levels of pLV-FVIII injected rats (n = 4) were found to be significantly increased up to 5.19 ± 0.14 ng/mL at post-injection 1st week (vs. 0.21 ± 0.05 ng/mL in pLV-LacZ injected rats, p < 0.05). These elevated levels were maintained for 4 weeks (2.52 ± 0.83 ng/mL vs. 0.17 ± 0.08 ng/mL in pLV-LacZ injected rats rats, p < 0.05) (Fig. 3).
Presence of plasma anti-FVIII antibodies concentrations
Since the rats used in these experiments were immune-competent, we suspected whether the low levels of FVIII concentrations in the pLV-FVIII injected rats after 6 weeks were due to the production of neutralizing antibodies to human coagulation FVIII protein. Analyses for antibody against human FVIII showed that neutralizing antibodies developed in pLV-FVIII rats after 10 weeks, but not in control rats (Fig. 4).
DISCUSSION
Data in this study showed that a single administration of an advanced generation lentivral vector, carrying the human B-domain deleted FVIII cDNA, into the hind limb of skeletal muscle of immune-competent rats resulted in effective elevation of circulating human FVIII.
Hemophilia gene therapy requires the gene delivery system that is efficient, safe, non-immunogenic and permits long term FVIII expression in the circulation. Only three lentivirus out of nine wild types of HIV gene have recently been engineered as the new vectors to transfer genetic material and produce functional, non-replicating viral vector.10 The lentivirus vectors are well recognized for their ability to transduce proliferating and non-proliferating cells at a similar efficiency.11 Data in the current study have shown that the advanced generation lentiviral vectors in Hela cells, carrying a FVIII cDNA, were capable of producing high concentrations of active FVIII in vitro.
The human B-domain deleted FVIII was chosen in this study, because the B-domain structure does not require procoagulant activity in contrast to full-length of human FVIII cDNA which contains a domain structure of A1-A2-B-A3-C1-C2.16 The B-domain deleted hFVIII retains coagulant activity and is secreted from cultured cells more efficiently than with full length FVIII. Furthermore, it is less sensitive to degradation.17,18
The liver appears to be the appropriate primary target organ for transduction since FVIII is naturally synthesized in hepatocytes and hepatic sinusoidal endothelial cells.13 Despite the fact that FVIII is synthesized in the liver, its biologically active form can be synthesized in a variety of tissues. Therefore, the site specific expression is not required. Ogata, et al.19 reported that transduction of adipocytes with vectors carrying the human FVIII gene may be applicable for hemophilia A gene therapy. The subcutaneous adipose tissue is accessible for vector administrations and can safely be resected in the event of adverse reaction. Ishiwata et al.20 explored the possibility of increasing circulating FVIII by tranducing adeno-associated virus (AAV) vectors into skeletal muscles, and showed sufficient expression of FVIII in the skeletal muscles and proven that AAV vectors could produce FVIII in the skeletal muscles. In our study, FVIII concentration levels were monitored throughout the 12 weeks period of time after a single injection of FVIII-expressing LV into rat's skeletal muscle, and the results showed sufficient expression of circulating human FVIII. Therefore, this gene transfer approach to the skeletal muscle could be an effective alternative system for hemophilia A gene therapy.
The major concern in vivo gene therapy for hemophilia A is the host immune response towards the viral vector. It would be an obstacle if multiple injections are required to achieve long-term therapeutic FVIII levels. The level of plasma FVIII in rat was monitored in the present study, and we observed that the FVIII level was gradually decreased from 2.52 ± 0.83 ng/mL to 0.37 ± 0.24 ng/mL at the 6th week after LV transduction, whereas anti-FVIII antibodies at 0.08 Bethesda unit/mL in serum were detected by the 10th week after vector injection. Since animals used in this study were immune-competent, we could not rule out the possibility of neutralizing antibodies developed against FVIII. The present data showed that FVIII levels were initially high (5.19 ± 0.14 ng/mL) accompanied by non-detectable anti-FVIII antibodies. Production of neutralizing antibodies after 6 weeks appears to be inversely correlated with the coagulation FVIII levels in the plasma. We observed that the plasma FVIII level was reduced by the 6th week, followed by the detection of neutralizing antibodies at the 10th week. There were 4 weeks of window period between the appearance of neutralizing antibodies and the reduction of plasma FVIII. It is highly possible that silencing promoter in vivo might have occurred, subsequently leading to decrease of FVIII production, since the levels of anti-FVIII antibodies were minimal. However, the expression of transgene with LV can last for several months.21 Therefore, the possibility of silencing promoter seems to be unlikely to take place in our study. To circumvent the neutralizing antibody formation, some investigators employed tissue specific promoters instead of ubiquitous promoters or immune deficient cells. Moayeri, et al.22 reported that therapeutic levels of FVIII were detected in the plasma of the transplant recipients for over 6 months of time: In this study, the recipient mice received minimally myeloablative dose of total body irradiation in order to blunt the host immune response. Kang, et al.23 showed that intravenous administration of GP64-pseudotyped feline immunodeficiency virus vector conferred sustained FVIII expression in hemophilia A mice for several months (7 months) without developing anti-human FVIII antibodies and further resulted in partial phenotypic correction. The use of a liver-specific promoter and vector modifications facilitated hepatocyte transduction rate by approximately 20 percent. Shi, et al.24 used the platelet-specific glycoprotein IIb gene promoter to direct FVIII expression, and demonstrated that the expression of FVIII in platelets has the advantage of delivering clotting factors directly to the site of an injury, despite the presence of FVIII inhibitory antibodies. Therefore, the presence of pre-existing inhibitory antibodies against FVIII might not be a contraindication for such a gene transfer system. Liu, et al.25 used non-viral vectors to overcome immunological problems associated with viral vectors: They used Sleeping Beauty transposon-based delivery to target endothelial cells for the production of human FVIII and achieved therapeutic levels of FVIII and phenotypic correction of bleeding disorder in the animal model throughout 24 weeks period of time. The level of neutralizing antibodies against FVIII protein was negligible. To precisely understand the underlying antigen presentation in immunologic mechanisms, the gene transfer immune response and induction tolerance would be the key for the success in hemophilia gene therapy.
We expect that only extramuscular sites are transduced via transport through the bloodstream. To resolve problems, we may additionally need to perform a comparative study using an another animal model that is genetically closer to humans rather than rat.
In summary, the present study showed an elevated circulating FVIII level in rats by single administration of advanced generation LV carrying FVIII cDNA, and suggested that the gene transfer approach via skeletal muscle could be an effective therapeutic tool for hemophilia A.



 XML Download
XML Download