

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
CD4+ T helper (Th) precursor cells are activated by the antigenic stimulation of T cell receptor (TCR) and are subsequently differentiated into different subsets of effector Th cells in order to boost the immune responses.1 Th1 and Th2 cells are traditionally thought to be the major subsets generated upon antigenic stimulation and produce distinct cytokine interferon γ(IFNγ) and interleukin (IL)-4, which are then involved in the elimination of intracellular and extracellular pathogens, respectively.2 Coordinated cytokine signaling induces the activation of specific transcription factors to promote lineage-specific cytokine production. While T-box-containing protein expressed in T cells (T-bet) is activated by IL-12 and IFNγ and is exclusively expressed in Th1 cell differentiation,3 GATA-binding protein 3 (GATA-3) and c-Maf are required for the chromatin remodeling and direct activation of Th2 cytokines IL-4, IL-5, and IL-13 for Th2 cell development.4,5 The balance of Th1/Th2 cells is thought to be determined by the expression ratio of T-bet/GATA-3 and is important for inducing autoimmune and allergic immune responses.6 However, the Th1/Th2 paradigm was recently shifted to the Th1/Th2/Th17/regulatory T (T-reg) hypothesis, a multi-lineage commitment from the same Th precursor cells.7,8 regulatory T cells, referred to as regulatory T cells, express forkhead box P3 (FoxP3) and suppress activated immune responses by producing transforming growth factor β(TGFβ)9,10 whereas Th17 cells induce retinoic acid-related orphan receptor γt (RORγt)-mediated IL-17 production and control the inflammatory autoimmune response.11,12 The differentiation of Th17 and T-reg cells requires the activation of TGFβ-mediated signaling, and IL-6 selectively drives Th17 cell differentiation from TGFβ-stimulated Th cells by promoting sequential activation of IL-21 and IL-23 signaling.10,13
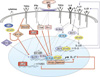
Here, we review the current understanding of the transcription factors involved in the regulation of Th17 cell differentiation, including updates of RORγt, FoxP3, and other Th17-specific transcription factors such as interferon regulatory factor 4 (IRF4), B-cell activating transcription factor (BATF), peroxisome proliferator activated receptor (PPARγ), T-bet, and suppressors of cytokine signaling (SOCS) 3 (Fig. 1).
TGFβ- AND IL-6-MEDIATED TH17 CELL DIFFERENTIATION
Many scientists have reported that TGFβ and IL-6 are essential for Th17 cell differentiation.12,14,15 TGFβ, which is produced by innate immune cells, binds to its specific receptor and induces engagement of TGFβ receptor I and II with subsequent activation of receptor-associated SMADs. Activated SMADs interact with a variety of transcription factors, resulting in chromatin remodeling and modulation of gene transcription of TGFβ target genes.16 TGFβ inhibits signal transducer and activator of transcription (STAT5)-mediated IL-2 production in TCR-stimulated T cells17 and also interferes with Th1 and Th2 cell differentiation by inhibiting expression of master transcription factor T-bet and GATA-3.18,19 In addition, TGFβ increases FoxP3 expression and induces generation of T-reg cells.10 FoxP3-positive T-reg cells potently suppress cell proliferation and differentiation of Th cells by boosting TGFβ production.10 Overexpression of TGFβ in a T cell-specific manner in mice leads to the generation of T cells with regulatory functions and protects IL-2-deficient mice from developing severe systemic inflammation with autoimmunity.20,21 However, additional production of the pro-inflammatory cytokine IL-6 along with TGFβ suppresses FoxP3 expression and T-reg cell generation and simultaneously induces IL-17 production, resulting in Th17 cell differentiation.12 Consistent with this, TGFβ transgenic mice treated with myelin oligodendrocyte glycoprotein (MOG) in complete Freund's adjuvant (CFA) exhibit substantially increased Th17-mediated immune responses and aggravated experimental autoimmune encephalomyelitis (EAE),12 indicating that TGFβ plus IL-6 induces the generation of Th17 cells. Despite the importance of TGFβ function in murine Th17 cells, TGFβ is dispensable for human Th17 cell differentiation. Instead, human Th17 cells are induced by stimulation with IL-6, together with another cytokine such as IL-1, IL-21, or IL-23.15,22,23
STAT3 AND RORγt AS KEY TRANSCRIPTION FACTORS FOR TH17 CELL DIFFERENTIATION
IL-6 is a key factor for inducing human and murine Th17 cell differentiation by activating STAT3 and RORγt expression. STAT3 activation is induced not only by IL-6 but also by IL-21 and IL-23.13,24,25 While STAT3-null cells fail to express RORγt and produce a diminished level of IL-17, retroviral restoration of STAT3 rescues the IL-17 defect.26 Although it is still unclear whether STAT3 modulates RORγt gene transcription, activated STAT3 directly binds to the STAT-binding sites in the IL-17 gene promoter and increases IL-17 gene transcription.27 In addition to the functional importance of STAT3 activation in Th17 cell differentiation, RORγt has been identified as a master regulator of Th17 cell differentiation. Analogous to STAT4-mediated T-bet in Th1 cells and STAT6-dependent GATA-3 in Th2 cells, Th17 cells require activation of STAT3 and subsequent RORγt induction.11 RORγt, a spliced isoform of RORγ, is a member of the nuclear receptor superfamily, and is closely related to the retinoic acid receptor (RAR) subfamily,28 and is required for thymocyte survival and lymphoid organogenesis.29 Deficiency of RORγt results in profound Th17 deficiency and protects mice from EAE development.11 Ectopic overexpression of STAT3 in RORγt-deficient cells, and vice versa, fails to restore IL-17 production.13 This suggests that STAT3 and RORγt may each regulate the other's gene transcription and induce IL-17 expression parallel to some extent. More recently, RORα was reported as a novel Th17-specific transcription factor. Like RORγt, RORα is induced by TGFβ plus IL-6 in a STAT3-dependent manner, and promotes Th17 cell differentiation through direct activation of IL-17 gene transcription.30 Double deficiency of RORγt and RORα results in complete blockade of IL-17 production and EAE development.30
IRF-4 FUNCTIONS IN IL-21-INDUCED TH17 CELL DIFFERENTIATION
IRF4 was originally identified as a GATA-3 inducer in Th2 cell differentiation.31,32 However, IRF4-null mice exhibit impaired generation of Th17 cells in response to TGFβ and IL-6 and increased resistance to EAE.33 In addition, IRF4-deficient Th cells exhibit an intrinsic defect in the autocrine IL-21 loop and an increased population of FoxP3-mediated T-reg cells with no effect on STAT3 activation and SOCS3 expression.34 A more recent report implies that IRF4 is activated upon IL-1 signaling and is critical for early Th17 cell differentiation.35 Despite the importance of IRF4 in Th17 cell differentiation, the molecular mechanisms are unclear. The fact that IRF4 interacts with NFATp to induce IL-4 expression may suggest that IRF4 modulates NFATp-dependent IL-2 expression, which is associated with IL-17 production.36,37
BATF AS A PROMOTER FOR TH17 CELL DIFFERENTIATION
The BATF is a basic leucine zipper (bZIP) transcription factor and dimerizes with Jun class factors of the activator protein-1 (AP-1) family.38-40 BATF is known to function as a potent inhibitor of AP-1-mediated gene expression via the phosphorylation of BATF.40-42 In addition, expression analysis reveals that BATF is highly expressed in hematopoietic cells and is increased in B and T cells by the activation of NF-κB in response to viral infection or IL-6-stimulation.43-47 BATF gene transcription is substantially increased in activated Th cells subsets including Th1, Th2, and Th17 cells.48 Despite its wide expression in all Th1, Th2, and Th17 cells, BATF-deficiency fails to generate IL-17 in CD4+ and CD8+ T cells in vitro and in vivo, but rather increases T-reg cell generation, thus protecting mice from EAE development.48 While the levels of RORα and RORγt expression are not sustained in BATF-deficient Th17 cells compared with those in wild type (WT) cells, enforced RORγt expression is not able to restore IL-17 production in BATF-deficient Th cells. Nevertheless, BATF synergizes with RORγt to induce IL-17 expression through direct interaction with the IL-17 gene promoter.48 Many questions regarding BATF, such as whether IL-6-induced STAT3 activation is affected by BATF deficiency and whether BATF is required for DNA binding of RORγt or IRF4, remain to be addressed in the future.49
FOXP3, T-BET, AND ETS-1 SUPPRESS TH17 CELL DIFFERENTIATION
Differentiation of FoxP3-directed T-reg cells and RORγt driven Th17 cells has been shown to be triggered by TGFβ signaling, but the Th17 differentiation program requires additional IL-6 or IL-21 cytokine signaling either to switch off FoxP3 or to switch on RORγt,10,11,15 suggesting reciprocal regulation of T-reg and Th17 cells during Th cell differentiation. It has been asked whether T-reg can be converted to Th17 in response to IL-6 and how FoxP3 and RORγt modulate each other's expression or activity.30,50,51 Interestingly, FoxP3 and IL-17 are both induced upon TGFβ stimulation.13,52 In addition, FoxP3-positive Th cells produce IL-17 in the presence of IL-6 through the activation of RORγt, whereas FoxP3 antagonizes RORγt activity in a manner dependent on SMAD4, suggesting the plasticity of T-reg cells.30 Others also report that FoxP3 inhibits IL-17 expression by antagonizing RORγt function in a TGFβ concentration-dependent manner53 or through direct interaction with RORγt.54 Like the suppressive function of FoxP3 on IL-17 expression, the Th1-specific transcription factor T-bet suppresses RORγt-mediated Th17 cell differentiation.55-57 Several functional studies indicate that T-bet suppresses RORγt expression and Th17 cell differentiation and further attenuates autoimmune responses.56,58-62 Nonetheless, the mechanism by which T-bet directly or indirectly inhibits IL-17 expression and whether T-bet antagonizes RORγt activity remain to be characterized. In addition, a T-bet-interacting transcription factor, Ets-1 positively modulates Th1 cell differentiation but inhibits Th17 cell generation.63,64 Ets-1-deficient Th cells exhibit preferential differentiation into Th17 cells and increased IL-22 and IL-23 receptor expression.64 Moreover, targeting of Ets-1 by microRNA miR-326 promotes Th17 differentiation.65 Since there is no apparent interaction between Ets-1 and IL-17 gene promoter, how Ets-1 modulates IL-17 expression must be defined in the future.
PPARγ AND E-FABP MODULATE TH17 CELL DIFFERENTIATION
Peroxisome proliferator-activated receptorγ (PPARγ) is a nuclear receptor like RORγt and RORα and forms heterodimers with retinoid X receptors (RXRs) to bind to the gene promoter.66,67 PPARγ activation upon ligand binding is critical for the expression of genes such as adiponectin and fatty acid-binding protein (FABP) (also referred as aP2) involved in adipocyte differentiation and lipid metabolism68,69 While enforced PPARγ expression induces adipocyte differentiation from fibroblasts, PPARγ-deficiency attenuates white adipose tissue development.70 Although PPARγ functions as a master transcriptional regulator for adipocyte differentiation, the anti-inflammatory activity of PPARγ is also well-characterized.71-73 The anti-inflammatory function of PPARγ is mediated through the inhibition of both maturation and function of dendritic cells and macrophages.74,75 More precisely, the ligand-binding domain of PPARγ is sumoylated upon ligand activation and prevents the removal of repressor complexes composed of nuclear receptor corepressor and histone deacetylase-3, thus resulting in sustained repressor complex-induced silencing of pro-inflammatory cytokine genes.76,77 In addition to the modulation of macrophage function, PPARγ modulates T cell activity by inhibiting IL-2 production in T cell receptor-stimulated Th cells78 and by suppressing Th2 cell differentiation.79 Therefore, PPARγ ligands including endogenous and synthetic agonists such as linoleic acid, prostaglandin J2, and thiazolidinediones have been extensively studied due to the interest in treating inflammatory diseases.71,80,81 A recent study demonstrates that PPARγ is an intrinsic suppressor for Th17 cell generation.82 PPARγ activation is thought to prevents removal of repressor complexes from RORγt gene promoter, thus suppressing RORγt expression and RORγt-induced Th17 cell differentiation in an intrinsic manner. Moreover, human multiple sclerosis patients are impressively susceptible to PPARγ-mediated suppression of Th17 cell development, strongly asserting PPARγ as a promising target for specific immunointervention in autoimmune disorders.82
In contrast to the suppressive function of adipogenic PPARγ, epidermal FABP (E-FABP) is characterized as a positive modulator of IL-17 production in Th cells.83 FABP-deficiency contributes to the protection from EAE development,84 which has been explained by the reduced level of pro-inflammatory cytokines in macrophages and dendritic cells.85 Moreover, FABP-deficient Th cells express increased amounts of PPARγ and subsequently suppress IL-17 production; however, this can be reversed by treatment with the PPARγ antagonist, GW9662.83 More detailed molecular mechanisms of E-FABP have yet to be characterized.
OTHER TRANSCRIPTIONAL MODULATORS FOR TH17 CELL DIFFERENTIATION
The SOCS inhibit STAT-mediated cytokine signaling.86,87 Since SOCS1 suppresses both IFNγ- and IL-4-mediated Th1 and Th2 cell differentiation, genetic ablation of SOCS1 results in unconditional hyperactivation of T cells.88 In addition, T cell-specific SOCS1-conditional knockout mice exhibit attenuated Th17 cell generation and induced hyperactivation of SOCS3 in Th cells,89 suggesting that SOCS1 as a transcriptional activator for Th17 cell development. Activated SOCS3 is known to selectively suppress STAT-3 activation induced by IL-6, granulocyte-colony stimulating factor (GCSF), and leptin,90,91 whereas deficiency of SOCS3 increases TGFβ production and simultaneously enhances Th17 cell development.27,92 It is also reported that TGFβ inhibits SOCS3 gene transcription and prolongs STAT3 activation during Th17 cell differentiation.93
CONCLUSION
With the recent reports of the functions of IL-17-producing Th17 cells in autoimmune responses and the regulatory mechanisms for Th17 cell differentiation, a new paradigm of Th cell differentiation has been established. The Th1/Th2 paradigm is mainly shifted to a Th1/Th2/ Th17/T-reg program. This review describes the function and potential mechanisms of transcription factors critical for the regulation of Th17 cell differentiation and also includes interconnection among transcription factors such as Th1-specific T-bet, T-reg-limited FoxP3, and Th17-specific RORγt . These transcription factors have prevalent roles in determining lineage commitment through interaction with cytokine gene promoters and/or other lineage-specific transcription factors. Targeting these transcription factors, as well as signature cytokines, may be beneficial for controlling several autoimmune diseases, although research is currently underway to identify the detailed mechanisms.
 XML Download
XML Download