

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Helicobacter pylori (H. pylori), a gastric pathogen in humans, survives intragastric acidity long enough to colonize the stomach. The gastric pH in the mucus layer is thought to vary between 4 and 6.5, with occasional acid alterations to less than pH 2.1-3 Resistance of H. pylori to acid alterations requires production of ammonia by urease-mediated degradation of urea. H. pylori produces large amounts of urease that account for up to 10% of its total protein content.4,5 Early in this field of study of H. pylori, the presence of surface urease was suggested to produce a cloud of ammonia that neutralizes the bacterial environment,6,7 however, intrabacterial urease activity was thought to play only a minor role in acid resistance. This concept is now known to be incorrect for many reasons that have experimentally been established in several laboratories. The evidence from earlier studies revealed that external urease results from cell lysis, and that the surface-bound urease is too low to contribute to acid resistance.8 The most likely explanation for the increase of urease activity observed with acidification is that there is a urea transporter in the bacteria.9 The urease gene cluster has seven genes: UreA, UreB, UreI, UreE, UreF, UreG, and UreH. UreI encodes an inner membrane protein with six transmembrane segments and is likely to be the transporter that is acidactivated, enabling urea access to the intrabacterial urease. With activation, there is a large increase of urea entry, thus increasing the intrabacterial urease. The increased urea uptake is energy-independent, temperature-independent and nonsaturable, indicating that UreI is a urea channel. Activation of this channel is rapid and is responsible for acute regulation of the ability of the bacterium to buffer its periplasmic pH.10
Although severe acid alterations have been shown to depend on urease activity, relatively little is known about the mechanisms involved in the growth at mildly acidic conditions. H. pylori ureI mutants were not able to elevate their urease activity in a medium at pH 4.011; acid-induced increase of urease activity was entirely dependent on UreI at pH lower than 4.0. However, elevated levels of urease activity were observed in mutants at pH between 4.0 and 5.5. These findings suggest that ureI independent mechanisms are sufficient for the maintenance of bacterial viability in such media in the presence of urea. During growth at acidic pH, urease-independent acid resistance mechanisms are essential,12 and 10 loci, not related to urease, are required for this process.13 One of these is the iron-responsive regulator Fur, which is responsible for iron homeostasis in many bacterial species.14 This repressor down-regulates transcription of iron transport systems when the intracellular concentration of Fe2+ exceeds a certain level. Fur is known to be involved in the acid resistance of several bacteria.15 Bijlsma, et al.16 reported that growth of fur mutants was severely impaired under acidic conditions. Addition of extra iron or removal of iron from the growth medium did not improve the growth of the fur mutant at an acidic pH, indicating that the phenotype of the fur mutant at low pH was not due to increased iron sensitivity. In their study, the urease activities of wild-type and fur mutant H. pylori were comparable, and addition of urea to the growth medium at pH 5 restored growth of the fur mutants to levels similar to that of the wild-type strain, indicating that the role of Fur in acid resistance of H. pylori is independent of urease. Fur was demonstrated to mediate iron-responsive regulation of the H. pylori paralogous amidases AmiE and AmiF.17 Both amidases degrade amide substrates to ammonia and corresponding carboxylic acid; they probably represent alternative systems for ammonia production at times when urea is in a short supply.5,18-20
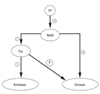
NikR is the regulator of nickel uptake. In H. pylori, nickel metabolism has been of interest, because of its role as a cofactor of the important colonization factor urease. The NikR is known to directly control urease expression and regulate the uptake of nickel; it is also able to regulate the expression of other regulatory proteins including Fur.5,21 Accordingly, van Vliet et al. hypothesized a cascade where fur controls the amidases independently of urease, and NikR regulates Fur and urease (Fig. 1). However, there have been no other studies targeting the putative link bet-ween urease and Fur without using the measurement of ammonia production for the determination of urease activity.
In this study, we observed for the first time that Fur influences urease activity, using the radiometrical measurement for urease activity of intact H. pylori at different pH, and the real time PCR of wild and fur mutant strains of urease genes.
MATERIALS AND METHODS
Bacterial strains and culture conditions
The H. pylori strain ATCC 43504 was obtained from the American Type Culture Collection. An ATCC 43504 fur deletion mutant was constructed by allelic exchange using a kanamycin resistance gene as described below. H. pylori was grown under microaerobic conditions (5% O2, 10% CO2, 85% N2) either on trypticase soy agar (TSA) plates supplemented with 5% sheep blood (Becton Dickinson, Cockeysville, MD, USA) or in brain heart infusion (BHI) medium (Difco Laboratories, Detroit, MI, USA) supplemented with 7% horse serum (Gibco BRL-Life Technologies Inc., Carlsbad, CA, USA) and 0.25% yeast extract (Difco Laboratories). All bacteria were grown in the media with the Dent selective supplement (Oxoid Limited, Hampshire, UK), and the fur mutant was grown in the presence of 20 µg/mL of kanamycin (Sigma Chemical Co., St. Louis, MO, USA).
Construction of the fur knockout mutant
A genomic knockout of HP1027 was constructed by homologous recombination (fur knockout mutant). pBluescript (Stratagene) containing a kanamycin resistance gene in the multicloning site flanked by SalI (5') and BglII (3') was used to generate the knockout plasmid. Primers were designed to flank the regions approximately 600 bp upstream of the 5' end of the gene and 400 bp downstream from the 3' end. The upstream segment was amplified with a 5' primer containing a site for digestion by XbaI (5'-GCTTCCATC TAGAATCTGGCGCGCTTGATTGC-3') and a 3' primer containing a site for digestion by SalI (5'-GTTTCTAATG TCGACATGCTGATATCTTCC-3'). The downstream segment was amplified with a 5' primer containing a site for digestion by BglII (5'-CCAAGCTG ATTAGATCTG ACATGAAAATGTTTGTGTGG-3') and a 3' primer containing a site for digestion by Acc65I (5'-GCATAATG GTACCTTCTATGCTTATGG-3'). The purified PCR products were sequentially ligated into pBluescript around the kanamycin resistance gene. The construct was introduced into the H. pylori strain ATCC 43504 by natural transformation, and colonies were selected in the presence of kanamycin (20 µg/mL). The knockouts were confirmed by a series of PCRs.
Survival assay
The H. pylori wild type and the fur mutants were grown overnight on agar plates. The bacteria were removed from the plates and resuspended in 10 mL of BHI medium, pH 7.4 or 4.0, and incubated for 60 min in a microaerobic environment at 37℃ Tenfold serial dilutions of the bacterial suspension were plated on TSA plates supplemented with 5% sheep blood and incubated for 3 to 5 days in a microaerobic atmosphere at 37℃. Survival at pH 4.0 was determined for each condition by comparison to the pH 7.4 controls. All experiments were performed in triplicate.
Membrane integrity
The integrity of the inner membrane of the bacteria was evaluated by confocal microscopy using membrane-permeant and -impermeant fluorescent DNA probes (Live/Dead; Molecular Probes). A compromised inner cell membrane was considered indicative of cell death. The membrane-permeant fluorescent DNA probe SYTO9, when bound to DNA and excited at 488 nm, emits a green light (emission, > 505 nm). Propidium iodide, which is membrane impermeant, emits a red light (excitation, 543 nm; emission, > 633 nm) when bound to DNA and quenches SYTO9 fluorescence. Therefore, viable bacteria will emit a green light but bacteria with their inner membrane integrity compromised will fluoresce red.
Wild-type H. pylori and the fur mutants were grown overnight on TSA plates at 37℃ in a microaerobic atmosphere. The bacteria were scraped off of the plates and suspended in BHI medium at pH 7.4 or 4.0 containing the Live/Dead indicators. The bacteria were incubated for 30 min at 37℃ in a microaerobic environment at the two pH values and then mounted on glass slides. Fluorescence was observed by confocal microscopy (LSM 510; Carl Zeiss, Inc., Oberkochen, Germany).
Urease activity of intact H. pylori between pH 3 and 8
Urease activity was measured radiometrically.8 Wild-type and fur mutant bacteria were added to 100 mM sodium phosphate buffer containing 5 mM KCl, 138 mM NaCl, 0.5 mM MgCl2, 1 mM CaCl2, 10 mM glucose, 1 mM glutamine, and 5 mM [14C]urea with a specific activity of 10 µCi/µmol at pH 3, 4, 5, 6, 7, and 8. The range of pH of the buffers used was between 4.0 and 8.5. The pH of the buffer, between 4.5 and 8.5, was achieved by mixing various amounts of 100 mM monobasic and dibasic sodium phosphate to the desired pH. Below pH 4.5, the desired pH was achieved by the addition of HCl. The pH of the buffer during the course of the experiment did not change by more than 0.1 pH unit, as measured with a pH electrode. Plastic wells containing 0.5 M KOH-soaked filter paper hung from rubber stoppers were used to collect the liberated 14CO2 that resulted from the hydrolysis of urea by urease. Urease activity was measured for 30 min at 37℃ with constant agitation. The reaction was terminated by the addition of 5 N H2SO4, which liberates all labeled CO2 from the incubation medium. The wells containing the filter paper were placed in a scintillation cocktail (HiIonicFluor; Packard Instruments, Meriden, CT, USA), and the radioactivity was measured by scintillation counting (1216 RackBeta; LKB Instruments, Gaithersburg, MD, USA). All experiments were performed in triplicate. The urease activity is reported as micromoles of CO2 released per minute per milligram of protein. The protein concentration was determined by the Lowry method.
Real-time qPCR
Unique primers were designed for 100- to 300-bp regions of five urease genes (ureE, ureF, ureG, ureH, ureI). Primer design was aided by Primer 3 software available at http://www-genome.wi.mit.edu/genomesoftware/other/primer3.html.22 Genomic DNA from the H. pylori strain ATTC 43504, serially diluted 10-fold, was used to obtain the standard curve. Each reaction mixture, which contained the SYBR Green label (Qiagen Inc., Hilden, Germany) and the primers, was added to a 96-well plate along with 1 µL of cDNA or genomic DNA template; the final reaction volume was 50 µL/well. The negative control contained the reaction mixture but no DNA. The reactions were performed with an Icycler real-time PCR machine (Bio-Rad, Hercules, CA, USA) for 40 cycles (95℃ for 30 s, 52℃ for 30 s, 72℃ for 40 s). Data were collected during the extension step and were expressed in arbitrary fluorescence units per cycle. A melting curve was used at the end to confirm that there was only one peak and only one product. Each sample was measured in duplicate.
RESULTS
Deletion of H. pylori fur (HP1027)
The H. pylori fur deletion mutant was constructed by homologous recombination. The gene deletion was confirmed with a series of PCRs using primers to the kanamycin resistance cassette and the upstream sense and downstream antisense primers used to construct the original knockout plasmid. The kanamycin resistance cassette used was 844 bp, from pUC4K (Amersham), and its presence was confirmed in the genomic DNA of the knockout. The remaining primers were used in combination with the kanamycin resistance gene primers to confirm that the resistance gene was in the proper position.
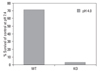
Survival of H. pylori fur mutant and wild strain at pH 4.0 or pH 7.4
Wild-type H. pylori and fur mutants were compared for survival. Both strains of H. pylori were incubated at pH 4.0 and pH 7.4 for 1 hour and bacterial survival was determined by colony counting. The survival of wild type H. pylori at pH 4.0 was 71.1% of the control at pH 7.4, whereas only 2.9% of H. pylori fur mutants survived at pH 4.0 (Fig. 2).
Urease activity of intact H. pylori between pH 3 and 8
The urease activity curve of the intact fur mutants showed the same shape as the wild-type but was approximately 3-fold lower than the wild-type at a pH less than 5 (Fig. 3).
Real-Time qPCR
Real time PCR, of both strains, was performed for the urease genes: ureI, ureE, ureF, ureG, and ureH. Cycle thre-shold values were calculated and converted into RNA concentration using the statistical standard curve. The expression of all genes was consistently down-regulated in the fur mutants (Fig. 4). RNA concentration was approximately 2-fold lower in ureE, 3-fold lower in ureF, 5-fold lower in ureG, 2-fold lower in ureH, and remarkably 20-fold lower in ureI, the actual urea channel.
DISCUSSION
The results of this study demonstrate that Fur is involved in acid resistant H. pylori urease activity contrary to van Vliet's hypothesis that fur controls the amidases independently of urease (Fig. 1). This was strongly supported by our findings that even though the curves of urease activity, in the intact H. pylori strains, have the same pattern, the strength of the activity was significantly different at a pH under 5, and that the expression of the urease genes was down-regulated in the fur mutants.
The adaptation of H. pylori to growth at an acidic pH is multifactorial. Urease has been shown to play an important role in this process; animal models have demonstrated that urease-negative mutants of H. pylori are unable to colonize the stomach.23 There is also a urease-independent acid resistance in H. pylori; this is observed when H. pylori are grown under acidic conditions in the absence of urea.13 The iron-responsive regulator Fur is involved in acid resistance independent of urease. The NikR protein can control the expression of different pathways for ammonia production directly via urease and indirectly through the Fur regulator and possibly other regulators.21 In addition, growth at acidic pH induces changes in the lipopolysaccharide composition,24 increases the expression of chaperone-like proteins25 and results in the induction of ammonia-producing pathways.5
The growth defect of the fur mutant at low pH is consistent with the findings of a previous report16 that suggested fur mutants are acid sensitive. It is known that the role of Fur in acid resistant H. pylori is independent of urease. Originally, H. pylori Fur was implicated in the regulation of urease expression,26 and the acid sensitivity of the fur mutant were thought to be due to reduced urease activity. However, the urease activity of the wild strain and fur mutant proved to be comparable.16 In addition, the amidases, degrading amide substrates to ammonia, were shown to represent alternative systems for ammonia production in times of urea shortage.5 Most reports showing that Fur is independent of urease have measured ammonia production from urea hydrolysis by the Berthelot reaction for urease activity. However, we used a radiometrical measurement of urease activity. Our results revealed that the urease activity of the intact bacteria had the same pattern with a 3-fold difference observed when the wild strain and the fur mutants were compared. This finding would not be predicted from van Vliet's hypothesis that Fur and urease act independently.21 Therefore, we performed real time PCR of the urease genes: ureI, ureE, ureF, ureG, and ureH in order to confirm the urease activity results. The findings showed that the expression of the genes in the fur mutant was consistently down-regulated. This finding strongly supports our hypothesis that Fur is involved in the acid resistant H. pylori urease activity. Because UreI is an actual urea channel, which allows extremely fast transport of urea across the inner membrane of the bacteria,9 these results strongly suggest that Fur is related to the urease activity.
Three H. pylori regulators have been implicated in acid adaptation27: the NikR protein, the Fur protein and the HP0164/0165-HP0166 two-component system. Among these, the Fur protein has been suggested to be located between NikR and the HP0166 system within a cascade, NikR > Fur > HP0166.21 Fur appears to have various roles in H. pylori that have not been previously described. The role of Fur in acid resistant H. pylori appears to be inde-pendent of its role in iron acquisition.11 However, the biology of iron is closely related to acidity, because it is more soluble under acidic pH conditions. To elucidate the mechanisms by which Fur influences urease and acid resistance in H. pylori, further investigation of Fur in iron homeostasis and its association with gastric acidity is needed.


 XML Download
XML Download