

 PDF
PDF ePub
ePub Citation
Citation Print
Print


THE KEY PATHWAYS FOR DNA DAMAGE RESPONSE
The replication of mammalian cells is a high-fidelity process that assures an accurate passage of genomic information to the daughter cells. However, their genome is constantly challenged by endogenous metabolic byproducts and environmental factors that lead to the improper presence of single-strand DNA breaks (SSBs) and/or double-strand DNA breaks (DSBs).
Among all the types of damage, DSBs pose the greatest challenge to cells, and are dangerous and potentially lethal lesions.1 The DNA-damaging agents such as ionizing radiation, reactive oxygen species can cause DSBs in the cells.2 A DNA replication fork that encounters DNA single-strand breaks or other types of lesions will also produce DSBs. They can also occur at the ends of chromosomes due to a defective metabolism of telomeres.3,4 In addition, DNA DSBs can form in a programmed manner during development.5,6 Those breaks are generated to initiate recombination between homologous chromosomes during meiosis and occur as intermediates during developmentally regulated rearrangements, such as V(D)J recombination and immunoglobulin class-switch recombination. For a cell, to preserve genome integrity, DSBs need to be repaired promptly, and the repair can be fulfilled through two independent but not mutually exclusive mechanisms initiated by DDR: error prone non-homologous end joining (NHEJ) or relatively error-free homologous recombination (HR).1
In the host, DNA damage response (DDR) pathways are activated to respond to DNA breaks to maintain genomic integrity. The DNA damage response involves the sensing of DNA damage followed by transduction of the damage signal to a network of cellular pathways including cell cycle checkpoints, DNA repair, and the apoptotic pathway. In general, the DDR network consists of two major parallel pathways that respond to SSB or DSB.7-10 In this network, two phosphatidylinositol-3-related kinases, ataxia telangiectasia mutated (ATM) and ATM-Rad3-related (ATR), which are located at the top of the signal cascades, phosphorylate and activate a variety of molecules to execute the DNA damage responses.7-10 ATM is activated primarily by double-strand breaks induced by ionizing irradiation and acts during all phases of the cell cycle, whereas the ATR pathway responds to agents interfering with functions of DNA replication forks, such as ultraviolet light and hydroxyurea.7,11 ATM can also activate many of the downstream targets of the ATR pathway, indicating that the two major pathways of the DDR are interlaced in a cell.
ATM and ATR are proximal kinases that act as the core sensors and are central to the entire DDR. These two kinases, by collaborating with other sensor molecules, function to detect various forms of damaged DNA and trigger DNA damage response cascades. For example, the complex of MRE11-Rad50-NBS1 (MRN) is a major sensor of broken DNA that recruits ATM to the break sites.12-14 Several other proteins such as 53BP115,16 and MDC117,18 also participate in this process to locate ATM or facilitate activation of ATM at the lesion sites of the DNA. ATM and ATR activation lead to cell cycle arrest or checkpoint via phosphorylation of ChK1 and ChK2.10 For instance, ChK1 is phosphorylated by ATM/ATR at Ser345 and Ser317 in response to various types of DNA damages which can be induced by ionizing radiation, ultraviolet, hydoxyurea, and topoisomerase inhibitors,19-24 ChK1, in turn, phosphorylates the phosphatase CDC25A,22,25 which facilitate the degradation of CDC25A.26 As a result, CDC25A can no longer dephosphorylate and activate CDK2 and CDK1, causing cells arrested in late G1, S, and G2 phases.26,27 In parallel, ChK2 is a key kinase downstream of ATM, which is primarily involved in the DDR-induced apoptosis.10
The effectors of the DDR include proteins that participate in DNA repair, transcription regulation, replication, cell-cycle control, and apoptosis, such as CDC25, p53, and various DNA repair proteins such as BRCA1, BRCA2, and RAD51.8,28 Most of these effectors are downstream molecules of either ATM/ATR and/or ChK1/ChK2. The ATM signaling pathway can activate and recruit BRCA1/2 proteins to the DNA damage sites. BRCA1/2 induced double-stranded repair of breaks using HR through interaction with RAD51, which is an important HR DNA repair enzyme.29 p53 is another protein playing a pivotal role in the execution of the DDR.30 p53 can be phosphorylated and stabilized by ATM/ATR and by another PIKK family member, DNA-PK. p53 can also be phosphorylated by ChK2, which itself functions as a substrate of ATM in response to DNA damage.9,31 The phosporylation of p53 by these DDR kinases lead to reduced binding of p53 to MDM2, which in turn allows the replacement of ubiquitin moieties by acetylation, resulting in p53 stabilization and full activation.32 Upon activation, depending on the severity of the stress signals, the pattern of post-transcriptional modifications and the cellular context, p53 acting as a potent transcriptional activator and/or repressor can induce cell cycle arrest and/or apoptosis,30,33-36 and remarkably p53 has also been shown to directly localize to sites of DNA damage and promote proper repair.37
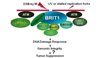
Recently, BRIT1/MCPH1 has been identified as a novel key DDR protein via the regulation of ATM/ATR pathways, modification of the chromosome structure, and direct involvement in DNA repair (Fig. 1). In this review, we will summarize the novel roles of BRIT1 in DDR, describe the relationship of BRIT1 deficiency with cancer development, and also discuss the use of novel synthetic lethality approach to target cancers with HR defects due to BRIT1 deficiency.
BRIT1 FUNCTIONS AS A NOVEL KEY DNA DAMAGE RESPONSE PROTEIN AT MULTIPLE LEVELS
Introduction of BRIT1
BRIT1 is a gene initially identified by a functional genomic screen as a novel repressor of hTERT.38 The amino acid sequence of BRIT1 was later matched to a putative disease gene called microcephalin (MCPH1). Mutations of this gene are found in a human genetic disease, primary microcephaly, which is characterized by a brain size one third of normal.39
BRIT1 is ubiquitously expressed in humans with higher levels observed in the brain, testes, pancreas, and liver. Human BRIT1 protein contains 835 amino acids with about 110 kDa of the molecular weight. According to MotifScan prediction, BRIT1 has three breast cancer carboxyl terminal (BRCT) domains, one nuclear localization signal motif, and a large central IMPDH domain. The BRCT domains of BRIT1, one in N-terminus (N-BRCT), the other two tandemly arranged in C-terminus (C-BRCTs) specifically bind to the phosphorylated proteins commonly involved in DNA damage response pathways. BRCT domains are conserved in BRCA1, MDC1, NBS1, and other important molecules involved in DNA damage signaling, DNA repair, and tumor suppression.40
The N-BRCT is required for centrosomal localization in irradiated cells, and also essential to rescue the premature chromosome condensation in BRIT1-deficient cells.41,42 In addition, N-BRCT is required for BRIT1's binding to SWI/SNF, a complex for regulation of chromosome relaxation.43 The C-BRCTs direct self-oligomerization of BRIT1, and are necessary for ionizing radiation-induced foci formation.41,44 The C-BRCT is also required for interaction and recruitment of BRCA2/RAD51 to the damage sites for execution of HR DNA repair function.45,46 The function of IMPDH domain predicted by MotifScan is not clear. However, the region (residues 376-485) in the central IMPDH domain (or middle domain), binding with Condension II, participates in homologous recombination.42
Role of BRIT1 in cell cycle control
BRIT1 has been demonstrated to regulate the expression of BRCA1 and Chk1, and it is required for the activation of the intra-S and G2/M cell cycle checkpoint after cellular exposure to ionizing radiation. In the absence of BRIT1, BRCA1 and ChK1 expressions are significantly reduced and NBS1 fails to be phosphorylated, leading to a loss of intra-S and G2/M checkpoint control.47,48 Cells derived from a microcephaly patient (BRIT1 defective) maintain a persistent level of cdc25A and a reduced level of Cdk1-clyclin B complex, both of which attributes to the entry of mitosis. Besides expression control of ChK1 and BRCA1, BRIT1 prevents premature entry into mitosis in an ATR-dependent and ATR-independent manner.49
BRIT1 function in DNA damage signaling as an early DDR mediator
BRIT1 can modulate activities of two distinct DNA damage repair networks, the ATM pathway and the ATR pathway. Upon exposure to DNA damaging reagents, BRIT1 co-localizes with numerous proteins associated with these two signaling pathways including γ-H2AX, MDC1, 53BP1, NBS1, p-ATM, ATR, p-RAD17, and p-RPA34.50 In the absence of BRIT1, all of these proteins, with the exception of γ-H2AX, fail to localize to sites of DNA damage. The depletion of BRIT1 inhibits the recruitment of phosphorylated ATM to double-stranded DNA break ends, and subsequently impairs the phosphorylation and recruitment of multiple down-stream members of the ATM pathway to the DNA damage sites including MDC1, 53BP1, and NBS1. BRIT1 deficiency also abolishes the UV-induced phosphorylation of RPA34 and reduces the levels of phosphorylated RAD17, indicating the roles of BRIT1 in the ATR pathway.50 All of these studies suggest that BRIT1 functions as a proximal factor in DDR pathway.
Using chicken cells as a study system, chicken BRIT1 can form irradiation-induced foci in ATM- and BRCA1-deficient cells, but not in H2AX-deficient cells.41 In addition, a study using the mouse embryonic cells (MEFs) with MDC1 or H2AX deficiency shows that BRIT1 foci formation can occur independently of MDC1 after DNA damage while it depends on H2AX phosphorylation.44 These observations support that BRIT1 acts early in DNA damage response pathways.
BRIT1's role in modifying chromosome structure
Chromatin remodeling is one of the fundamental mechanisms used by cells to relax chromatin during the process of DNA repair51,52 Our very recent study reports a novel function of BRIT1 as a regulator of ATP-dependent chromatin remodeling complex SWI/SNF in DNA repair.43 Upon DNA damage, BRIT1 increases its interaction with SWI/SNF through the ATM/ATR-dependent phosphorylation BAF170 subunit of this complex. This increase of binding affinity provides a means by which SWI/SNF can be specifically recruited to and maintained at DNA lesions since this interaction relaxes the chromatin structure and increases the access of the repair proteins, including RAD51, to the DNA damage sites.43 This chromatin remodeling function of BRIT1 may also contribute to the increased accessibility of many other DNA damage responsors, such as ATM, ATR, NBS1, MDC1, 53BP1, and RPA. Therefore, BRIT1 can function as a key molecule that links chromatin remodeling with DNA damage response in the control of DNA repair. In another attempt to identify BRIT1-associated proteins, it reveals that BRIT1 can physically bind to the Condensin II complex, which is involved in chromosome condensation. The interaction between these two proteins is required for BRIT1-mediated HR repair.42 BRIT1 can be indirectly involved in the DNA repair process via the regulation of the chromatin structure.
BRIT1's functions in HR and NHEJ DNA repair
HR and NHEJ are the two major pathways to repair DSBs.1 RAD51, a homolog of the bacterial RecA, is a central executioner in homologous recombination (HR), catalyzing the invasion of the single stranded DNA in a homologous duplex and facilitating the homology search during the establishment of joint molecules. A lack of BRIT1 can alleviate localization of RAD51 onto the DNA break sites.43,45,46 Thus, BRIT1 is critically implicated in the HR process. Using a HR reporter system, we and others have shown that HR repair efficiency is reduced in both human cells with BRIT1 depleted by siRNA and in MEFs isolated from BRIT1 knockout mice.42,43,45 In addition, NHEJ repair is impaired when BRIT1 is depleted. Thus, BRIT1 is required for both HR and NHEJ DNA repair.43
In addition to in vitro studies, the role of BRIT1 in HR DNA repair is clearly demonstrated using a BRIT1 knockout mouse model we generated recently.46 In mice, programmed DSBs are generated by SPO11 during meiosis for the initiation of meiotic recombination in spermatocytes. In response to these DNA damages, HR-DNA repair proteins such as RAD51 and BRCA2 are recruited to repair those SPO11-initiated DSBs, which ensures the proper process of meiotic recombination to produce sperm for reproduction. Interestingly, male BRIT1-/- mice are infertile with smaller testes and very few spermatids. BRIT1 deficiency does not impair spermatogonia or Sertoli cell proliferation. However, meiotic recombination in spermatocytes is impaired and meiosis is arrested at late zygotene of prophase I accompanying with apoptosis. In addition, RAD51/BRCA2 foci formation on the meiotic chromosome is abolished in BRIT1-/- mice, although DSB formation is not altered.46 Thus, BRIT1 is essential for HR DNA repair via recruitment of RAD51/BRCA2 to the DNA damaged sites.
Consistent with the role of BRIT1 in regulating the DNA repair function of BRCA2/RAD51, the meiotic phenotypes in BRIT1-/- mice are virtually the same as those observed in mice with a deficiency of BRCA253 and DMC1 (a homologue of RAD51).54,55 In these mice, spermatocytes are also arrested before or at the transition of zygotene to pachytene with aberrant chromosomal synapsis. In fact, like BRIT1-/- spermatocytes, BRCA2-/- spermatocytes also form DSBs without the consequent recruitment of RAD51 to the meiotic chromosome.53
A very recent report shows that in human cells, BRIT1 binds to the BRCA2/RAD51 complex and this binding is required for recruitment or retention of the BRCA2/RAD51 complex at the DNA repair sites.45 We also demonstrate that mouse BRIT1 can physically associate with RAD51 or BRCA2, and in the absence of BRIT1, recruitment of RAD51 and BRCA2 to chromatin is remarkably reduced while their protein levels are not altered.46 Thus, BRIT1 also functions directly in DNA repair by directing the recruitment of BRCA2/RAD51 to the DSBs.
BRIT1 DEFICIENCY, GENOMIC INSTABILITY, AND CANCER DEVELOPMENT
Genomic instability in BRIT1-deficient cells and mice
Due to its multiple functions in DDR, it is expected that BRIT deficiency would lead to genomic instability. Indeed, in human cancer cells, when BRIT1 is depleted by siRNA, these cells exhibit spontaneous chromosomal aberrations.50 The genomic instability induced by a loss of BRIT1 is also extensively studied using the BRIT1 knockout mouse.46 BRIT1-/- mice are more sensitive to irradiation with a shorter survival compared to the wild-type control mice. Mouse embryonic fibroblast (MEFs) isolated from the BRIT1-/- mice are also more sensitive to irradiation with severe chromosome breaks in response to irradiation.46 In addition, T cells isolated from BRIT1-/- mice exert more chromosomal aberrations as compared to the wild-types in the absence of any irradiation, indicating that BRIT1 plays a role in regulating spontaneous DNA damage.
BRIT1 deficiency in human cancers
In humans, BRIT1 is located at chromosome 8p23.1, where the loss of heterozigosity (LOH) is common in many types of cancer including breast and ovarian cancer. Recently, we have demonstrated that the levels of BRIT1 decreased in several types of human cancer.50 Using high-density array comparative genomic hybridization (CGH), we found substantial decreases in BRIT1 gene copy number in 35 of 87 cases (40%) of advanced epithelial ovarian cancer. Microarray data from a public database also showed that BRIT1 mRNA levels are markedly decreased in 19 of 30 cases (63%) of ovarian cancer specimens relative to BRIT1 mRNA levels in benign ovarian tissue specimens. Moreover, 72% of the 54 breast cell lines tested show decreases in the BRIT1 gene copy number. When comparing BRIT1 expression between non-transformed breast epithelial cells and established breast cancer cell lines, we also found significant decreases of BRIT1 RNA and protein expression in multiple breast cancer lines. In particular, we identified a BRIT1 gene deletion in one of the 10 cancer specimens analyzed which led to the loss of BRIT1 function in DNA damage response. Thus, BRIT1 is aberrant in human cancers and it suggests BRIT1 may function as a novel tumor suppressor gene.
BRIT1 deficiency and cancer development in mice
Genomic instability is observed in our BRIT1 knockout mice.46 Although BRIT1 knockout mice within one and half years old did not develop any tumor, when we crossed BRIT1 knockout mice to the p53 null background, we found a significant effect of BRIT1 deficiency in enhancing cancer susceptibility (unpublished data). Notably, our very recent preliminary data indicate that low dosage of irradiation can readily induce breast tumors in the mice with conditional knockout of BRIT1 in the mammary glands but not in the control littermates. It will be very interesting to investigate if, and to what extent, BRIT1 deficiency may contribute to the initiation and progression of cancer with the existence of oncogenic or genotoxic stress. For example, we can assess whether crossing an activated Ras or HER2 allele into the BRIT1 deficient background will result in increased genomic instability and tumorigenesis compared to either mouse strain alone. Thus, our BRIT1 null mouse will be a very valuable model for further assessing BRIT1's role in genome maintenance and tumor suppression in the future.
Perspective for targeting BRIT1 deficient-cancers with PARP inhibitors
DNA damaging agents, such as irradiation and chemodrugs (Doxorubicin, Carboplatin, and Cisplatin), are commonly used to treat cancer patients in clinics. However, the majority of DNA damage reagents are not specific and can cause toxicity in normal cells as well. Recently, based on the concept of synthetic lethality, poly (ADP-ribose) polymerase (PARP) inhibitors have been identified to kill BRCA1/2 deficient cells with high specificity, bringing great promise to the BRCA1/2 cancer patients.56-59 The underlying mechanism for this specific killing stems from a delicate synthetic lethal effect. PARP is an enzyme that facilitates repair of single-strand DNA breaks (SSBs). In normal cells, DNA damage generated by PARP inhibitors is well tolerated because of functional compensation from the HR-mediated DNA repair pathway.56,59 In contrast, HR-repair-defective cancer cells, such as BRCA1- or BRCA2-deficient cells, are unable to cope with this increased DNA damage and thereby exhibit hypersensitivity to PARP inhibitors.56-59 Given that the loss of BRIT1 impairs the BRCA2/RAD51 DNA repair function and leads to defective HR repair,42,43,46 it is possible that PARP inhibitors may serve as potent drugs specifically targeting BRIT1-deficient cancers. Recently, we performed an initial study and found that BRIT1-/- MEFs were more sensitive to PARP inhibitor olaparib as compared to BRIT1+/+ MEFs, suggesting that PARP inhibitors may be used to effectively treat BRIT1-deficient cancers. To further test this idea in the future, it is important to assess the response of the BRIT1-deficient cancers to potent PARP inhibitors using both cell culture and xenograft tumor models. It will also be worthwhile to test the effect of the combination of these PARP inhibitors with anti-cancer agents currently used in clinics to develop the optimal therapeutic remedies to target BRIT1-deficient cancers.
 XML Download
XML Download