

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemia-reperfusion (I/R) injury consists of inflammatory damage caused by the accumulation of free radical oxygen species, and triggers stress signaling processes that eventually result in cell death.1,2 Approaches to lessen I/R injury have been intensively studied, and the concept of ischemic preconditioning (IPC) has been introduced as an effective treatment for cardioprotection against I/R injury.3
IPC is an endogenous phenomenon whereby repeated brief episodes of coronary artery occlusion protect the heart against further prolonged ischemia.4 The cardioprotective effects of IPC have been demonstrated by a reduction in myocardial infarct size, improvement in postischemic contractile function, and reduction of arrhythmias.5
Anesthetic preconditioning (APC) by volatile anesthetics, morphine or propofol has been reported to protect I/R injury,6-8 similar to the effect of IPC such as blockade of Ca2+ overflow to the cytosolic space, an antioxidant-like effect, interference in the neutrophil/plate-endothelium interaction,9 and activation of ATP-sensitive potassium (KATP) channels.10
Intracellular Ca2+ overload, as a consequence of dysregulation of Ca2+ homeostasis, has been suggested to explain the adverse effects of I/R, which leads to cardiomyopathy and heart failure.11 Therefore, interruption of Ca2+ overload has been an important target for increased tolerance to I/R injury.
Thiopental, an ultrashort-acting barbiturate, has been used for anesthetic induction because of its rapid onset and short duration of action. It has been reported that thiopental depresses the release of reactive oxygen species (ROS) from neutrophils12 and inhibits lipid peroxidation as antioxidant properties.13 Thiopental is also primarily known as an anesthetic that protects the brain against I/R injury.14,15 Although thiopental reduces creatine kinase (CK) release, an indicator of cell damage, in isolated ischemic heart muscle16 and restores recovery of heart function at extremely high concentrations in the ischemic rat heart,17 its role in cardioprotection remains a matter of debate.
In cultured neonatal rat cardiomyocytes exposed to hypoxia and reoxygenation, several studies demonstrated that intermittent interruptions of reoxygenation with hypoxia after the hypoxic period (hypoxic postconditioning) reduces intracellular Ca2+ overload and prevents cardiomyocytes loss.18,19 Therefore, using primary cultured neonatal rat cardiomyocytes in the present study, we hypothesized that thiopental promotes protection against hypoxiareoxygenation injury by regulating Ca2+ homeostasis. Accordingly, we evaluated the proteins involved in maintaining Ca2+ homeostasis, apoptosis, and survival signals in order to investigate the effect on cardioprotection. This study provides an indirect model for the protection of the hearts from I/R injury.
MATERIALS AND METHODS
Animals and chemicals
Hearts of neonatal Sprague Dawley rats (1-2 days old) were used for isolation of cardiomyocytes. The experimental procedures were approved by the committee for the Care and Use of Laboratory Animals, Yonsei University College of Medicine.
Isolation of neonatal rat cardiomyocytes
Neonatal rat cardiomyocytes were isolated according to previously described methods.20 Briefly, hearts of 1-2 day-old Sprague Dawley rat pups were dissected and then the ventricles were washed with Dulbecco's phosphate-buffered saline solution (PBS) (pH 7.4, Gibco BRL, Carlsbad, CA, USA) lacking Ca2+ and Mg2+. Using micro-dissecting scissors, hearts were minced until the pieces approximately 1 mm3 and treated with 10 mL of collagenase I (0.8 mg/mL, 262 units/mg, Gibco BRL, Carlsbad, CA, USA) for 15 min at 37℃. The supernatant was removed and tissues were treated with fresh collagenase I solution for an additional 15 min. Cells in the supernatant were transferred to a tube with a cell culture medium [α-MEM containing 10% fetal bovine serum (FBS), Gibco BRL, Carlsbad, CA, USA]. Tubes were centrifuged at 1,200 rpm for 4 min at room temperature, and the cell pellet was resuspended in 5 mL of the cell culture medium. The above procedures were repeated 7-9 times until little tissue remained. Cell suspensions were collected and incubated in 100-mm tissue culture dishes for 1-3 hours to reduce fibroblast contamination. Non-adherent cells were collected and seeded to achieve a final concentration of 5×105 cells/mL. After incubation for 4-6 hours, the cells were rinsed twice with cell culture medium and 0.1 µM BrdU was added. Cells were then cultured with 10% (v/v) FBS in a CO2 incubator at 37℃ for 3-5 days.
Induction of hypoxia
Cardiomyocytes were exposed to increasing concentrations of thiopental (0.1-500 µM) and immediately replaced in the hypoxic chamber to maintain hypoxia. After 1 hour of exposure, the culture dish was transferred to the CO2 incubator and cells were incubated at 37℃ for 5 hours. Thiopental was diluted to various concentrations according to the described methods.21 Culture dishes containing cardiomyocytes in α-MEM were subjected to hypoxic stress in an anaerobic chamber (ThermoForma, Marietta, OH, USA) maintained at 37℃ in which ambient oxygen was replaced by a mixture of 5% CO2, 5% H2, and 90% nitrogen. Three mesh-encased wafers of desiccant, palladium catalyst, and activated charcoal further ensured anaerobic conditions by removing H2O, H2S, and H2O2.
Cell viability assay
Cell viability was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] assay. Cardiomyocytes were plated in triplicate wells in 96-well plates at a density of 1×104 per well, and incubated for 24 hours. The media of the samples were changed with degassed serum-free media, and samples were pretreated with various concentrations of thiopental (0.1-500 µM). Then, samples were subjected to a hypoxic chamber for 1 hour and exposed to reoxygenation at 37℃ for 5 hours. After the incubation period, MTT was added to each well to a final concentration of 0.5 mg/mL, and the cells were incubated at 37℃ for 3 hours to allow MTT reduction. Formazan crystals were dissolved by adding dimethylsulfoxide (DMSO) and the absorbance was measured at 570 nm with a spectrophotometer.
Immunoblot analysis
Cardiomyocytes that had been pretreated with 50 µM thiopental were subjected to hypoxic chamber for 1 hour and exposed to reoxygenation for 5 hours. Cells were washed once in PBS and lysed in lysis buffer (Cell Signaling, Danvers, MA, USA). Protein concentrations were determined using the Bradford protein assay kit (BioRad, Hercules, CA, USA). Proteins were separated in a 12% SDS-polyacrylamide gel and transferred to PVDF membrane (Millipore Co., Billerica, MA, USA). After blocking the membrane with Tris-buffered saline-Tween 20 (TBST, 0.1% Tween 20) containing 5% non-fat dried milk for 1 hour at room temperature, membranes were washed twice with TBS-T and incubated with primary antibodies for 1 hour at room temperature or overnight at 4℃. The following primary antibodies were used: rabbit anti-ERK, mouse anti-phospho ERK, mouse anti-Bcl-2, rabbit anti-Bax (Assay Designs, Ann Arbor, MI, USA), mouse anti-SERCA2a, mouse anti-NCX (Affinity Bioreagents, Rockford, IL, USA), and mouse anti-β actin antibodies (Sigma, St. Louis, MO, USA). The membrane was washed 3 times with TBS-T for 10 min, followed by incubation for 1 hour at room temperature with horseradish peroxidase (HRP)-conjugated secondary antibodies. After extensive washing, the bands were detected by enhanced chemiluminescence (ECL) reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The β-actin gene was used as the standard for equal of the protein samples. Band intensities were quantified using a Photo-Image System (Molecular Dynamics, Uppsala, Sweden).
Detection of apoptotic cells by TUNEL staining
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) analysis was performed with a commercially available kit according to the manufacturer's instructions (Intergen, Burlington, MA, USA). Cultured cardiomyocytes were prepared on coverslips in 24-well plates. Briefly, cells were fixed in 1% paraformaldehyde and pre-cooled ethanol: acetic acid (2 : 1) and equilibrated with an equilibration buffer. The samples were then treated with terminal deoxynucleotidyl transferase (TdT) and incubated in a humidified chamber for 1 hour at 37℃. After washing in PBS, anti-digoxigenin peroxidase conjugate was added, and incubation continued in a humidified chamber for 30 min at room temperature. The samples were washed with PBS, stained with peroxidase substrate (diaminobenzidine) for 5 min at room temperature, and observed by microscope. The apoptotic index (percentage of apoptotic nuclei) was calculated as apoptotic nuclei/total nuclei counted ×100%.
Caspase-3 assay
Relative caspase-3 activity was determined using the Apop-Target™ Capase-3 Colorimetric Protease Assay. This assay is based on the generation of free DEVD-pNA chromophore when the provided substrate is cleaved by caspase-3. Upon cleavage of the substrate by caspase-3, free pNA light absorbance can be quantified using a microplate reader at 405 nm. Briefly, after different treatments, the cultured neonatal cardiomyocytes (3×106) were harvested in lysis buffer (1M DTT), and cell extracts were centrifuged to eliminate cellular debris. Protein concentration was determined by the Bradford assay (Bio-Rad, Hercules, CA, USA). Aliquots (50 µL) of cell extracts were incubated at 37℃ for 2 hours in the presence of the chromophore substrate. Free DEVD-pNA was determined colormetrically. The comparison of absorbance of pNA from apoptotic samples with uninduced controls allowed determination of the fold increase in caspase-3 activity.
Confocal microscopy and fluorescence measurements
Cytosolic free Ca2+ was measured by confocal microscopy analysis. Neonatal rat cardiomyocytes were plated on glass coverslips coated with laminin (5 mg/cm2) for 1 day in cell culture medium (α-MEM containing 10% FBS) and 0.1 µM BrdU. Following pretreatment with 50 µM thiopental, cardiomyocytes were subjected to a hypoxic chamber for 1 hour and exposed to reoxygenation for 5 hours. After incubation, cells were washed with modified Tyrode's solution containing 0.265 g/L CaCl2, 0.214 g/L MgCl2, 0.2 g/L KCl, 8.0 g/L NaCl, 1 g/L glucose, 0.05 g/L NaH2PO4, and 1.0 g/L NaHCO3. Cells were then loaded with 5 mM of the acetoxymethyl ester of Fluo-4 (Molecular Probes, Eugene, OR, USA) for 20 min, in the dark and at room temperature, by incubation in modified Tyrode's solution. Fluorescence images were obtained using an argon laser confocal microscope (Carl Zeiss Inc., Oberkochen, Germany). This fluorochrome is excited by the 488 nm line of an argon laser and emitted light is collected through a 510-560 nm bandpass filter. Relative changes of free intracellular Ca2+ were determined by measuring fluorescent intensity.
RT-PCR analysis
Cardiomyocytes that had been pretreated with 50 µM thiopental were subjected to hypoxic chamber for 1 hour and exposed to reoxygenation for 5 hours. Cells were washed once in PBS. Total RNA was prepared by the UltraspectTM-II RNA system (Biotecx Laboratories, Inc., Houston, TX, USA) and single-stranded cDNA was then synthesized from isolated total RNA by avian myeloblastosis virus (AMV) reverse transcriptase. A 20-µL reverse transcription reaction mixture containing 1 µg of total RNA, 1X reverse transcription buffer (10 mM Tris-HCl, pH 9.0, 50 mM KCl, 0.1% Triton X-100), 1 mM deoxynucleoside triphosphates (dNTPs), 0.5 units of RNase inhibitor, 0.5 µg of oligo (dT)15, and 15 units of AMV reverse transcriptase was incubated at 42℃ for 15 min, heated to 99℃ for 5 min, and then incubated at 0-5℃ for 5 min. All primer pairs for DNA sequencing of genes related to Ca2+ homeostasis are shown in Table 1. The PCR condition was 94℃ for 3 min, 94℃ for 1 min, 41-49℃ for 1 min, and 72℃ for 2 min, 35 cycles, with a final extension for 10 min at 72℃. The GAPDH gene (primers 5'-ACCACAGTCCATGCCATCAC-3' and 5'-TCCACCACCCTGTTGCTGTA-3', 450 bp) was used as the internal standard. Signal intensity of the amplification product was analyzed using UVlband software (UVltec, Cambridge, UK).
Measurement of mitochondrial membrane potential
JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazoly-carbocyanine iodine) (Immunochemistry Technologies, LLC, Bloomington, MN, USA) was used to measure mitochondrial membrane potential (ΔΨm).22,23 At the end of the experimental procedure, cardiomyocytes were incubated with JC-1 (5 µmol/L) at 37℃ for 20 min in the dark. Flow cytometry analysis was performed on a FACSCalibur system (Becton Dickinson, Franklin Lakes, NJ, USA) using CellQuest™ software with 10,000 events recorded for each sample. JC-1 monomer (green) fluorescence was observed by excitation with the 488 nm laser and examination of the emissions at 530 nm. JC-1 aggregate (red) fluorescence was observed by examination of the emissions at 590 nm. Data was acquired in single parameter histogram with appropriate particle size and light scatter gating.
Statistical analysis
Data is presented as mean ± S.E.M. from more than 3 separate experiments performed in triplicate. Where results of blots and RT-PCR were shown, a representative experiment was depicted. Comparisons between multiple groups were performed with one-way analysis of variance (ANOVA) with Bonferroni test. Statistical significance was defined as p < 0.05 and p < 0.01.
RESULTS
Effect of thiopental on survival of hypoxia-reoxygenated cardiomyocytes
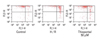
As shown in Fig. 1A, the viability of hypoxia-reoxygenated cardiomyocytes in the thiopental-treated group was greater than that of the untreated group. The concentration of thiopental largely influenced the survival of the hypoxia-reoxygenated cardiomyocytes, and 50-300 µM thiopental produced a significant increase in cell survival (p < 0.01). As the effects were similar from 50 to 300 µM, we used a concentration of 50 µM for the following experiments. In preliminary experiments, we tested the survival rate of hypoxia-reoxygenated cardiomyocytes following exposure to reoxygenation at each time period (30 min, 1 h, 2 h, 5 h, and 12 h), demonstrating that the best survival effect by thiopental was 5 hours (data not shown). Thus, we chose 5 hours for our experimental condition.
The activation of extracellular signal-regulated kinases (Erk 1/2) plays an important role in the mechanisms of cellular survival and proliferation through gene regulation.24 As Erk 1/2 are dual specificity kinases in mitogen-activated protein (MAP) kinase respectively, we examined phosphorylation of Erks (42 and 44 kDa) by immunoblot assay. The phosphorylating activity of Erks was dramatically lower in hypoxia-reoxygenated cardiomyocytes than in normal cells (p < 0.01). Additionally, cardiomyocytes pretreated with 50 µM thiopental showed higher levels of Erks phosphorylation than thiopental-untreated hypoxia-reoxygenated cells (p < 0.05), but the phosphorylation levels did not reach those of normal cells (Fig. 1B).
Effect of thiopental on activity of proteins related to apoptosis
To investigate the effects of thiopental on apoptosis in hypoxia-reoxygenated cardiomyocytes, we determined the expression of several proteins related to apoptosis. As shown in Fig. 2A, hypoxia and reoxygenation increased the expression of proapoptotic proteins, Bax, but decreased expression of the anti-apoptotic B cell leukemia/lymphoma-2 (Bcl-2) protein. When hypoxia-reoxygenated cells were treated with 50 µM thiopental, the expression level of Bax decreased, but Bcl-2 increased. The inhibition of apoptosis by 50 µM thiopental was confirmed by TUNEL analysis (Fig. 2B). Activation of caspase-3 during apoptosis has been linked to the proteolytic cleavage of cellular substrates and documented in the myocardium of end-stage heart failure.25 Hypoxia-reoxygenation stimulated the activation of caspase-3 in cardiomyocytes (p < 0.05), but the activity of caspase-3 was reduced when cells were treated with 50 µM thiopental (p < 0.05) (Fig. 2C).
Effect of thiopental on intracellular Ca2+ concentration
Exposure to hypoxia-reoxygenation increased intracellular Ca2+ level, but treatment with 50 µM thiopental restored the level to its baseline (p < 0.05) (Fig. 3).
Gene expression of proteins influencing Ca2+ homeostasis
To assess the expression of genes related to Ca2+ homeostasis, total RNA was isolated and analyzed by semiquantitative RT-PCR. We obtained a single band of an appropriate size. As illustrated in Fig. 4, when compared to normal cardiomyocytes, hypoxia-reoxygenated cardiomyocytes showed decreased expression levels of sarcoplasmic reticulum Ca2+-ATPase (SERCA2a), L-type Ca2+ channel, and sarcolemmal Ca2+ pump (PMCA1). In particular, the transcriptional levels of SERCA2a were much lower (p < 0.01). However, mRNA levels of Na+-Ca2+ exchanger (NCX) and calsequestrin increased in hypoxia-reoxygenated cardiomyocytes. Treatment of hypoxia-reoxygenated cells with 50 µM thiopental resulted in up-regulation of mRNA transcription levels of SERCA2a and PMCA1 (p < 0.05), and decreased NCX gene expression (p < 0.01). There were no significant changes in L-type Ca2+ channel and calsequestrin mRNA transcription levels when hypoxia-reoxygenated cells were treated with thiopental. In addition, phospholamban (PLB) and ryanodine receptor (RyR) expressions were not altered in hypoxia-reoxygenated cardiomyocytes treated with thiopental. To confirm functional changes of SERCA2a and NCX, Western blot analyses with anti-SERCA2a and anti-NCX antibodies were performed. As shown in Fig. 5, hypoxia-reoxygenated cardiomyocytes pretreated with 50 µM thiopental showed higher levels of SERCA2a (p < 0.05), but the expression of NCX protein decreased (p < 0.05). Therefore, these results are in agreement with changes in mRNA transcription levels.
Effect of thiopental on ΔΨm
To determine whether thiopental suppressed apoptosis through sustaining ΔΨm, neonatal cardiomyocytes were stained with JC-1. At higher potentials, JC-1 forms red fluorescenct J-aggregates (excitation/emission wavelength = 488/590 nm). When mitochondrial membrane is depolarized, JC-1 is a monomer and emits green fluorescence (excitation/emission wavelength = 488/530 nm).22,23 Percentage numbers in Q1, Q3, and Q2, Q4 represent proportions of cells with normal and depolarized mitochondrial membranes, respectively. Hypoxia-reoxygenation treatment increased mitochondrial membrane depolarization (Q2: 57.05, Q4: 9.91%), and this effect was reversed with 50 µM thiopental (Q2: 33.41, Q4: 6.70%) (Fig. 6).
DISCUSSION
In the present study, we observed significant reductions of hypoxia-reoxygenation induced apoptotic signals and intracellular Ca2+ content in thiopental-treated cells. In addition, thiopental activated proteins concerned with survival, attenuated alterations of genes involving Ca2+ regulation, and suppressed disruption of mitochondrial membrane potential were also observed. These results suggest that thiopental treatment protects neonatal rat ventricular cardiomyocytes against hypoxia-reoxygenation injury by inhibiting apoptosis through repression of intracellular Ca2+ overload. Our results also demonstrated that inhibition of Ca2+ overload is modulated by proteins involved in Ca2+ homeostasis. In the preliminary experiments, we have multiple criteria about reoxygenation time. The best survival effect by thiopental was 5 hours in neonatal cardiomyocytes. Thus, we chose that point (5 h) in our experimental condition.
Thiopental has been known to protect the brain against ischemic damage by attenuation of nitric oxide-induced cytotoxocity26,27 and enhanced glutamate release.28 It has also been shown that thiopental reduces anoxia-mediated Ca2+ influx in rat hippocampal slices.15 With respect to the effects on the heart, limited studies have reported that thiopental protects the heart against I/R-injury16,17 but it does not increase the cardioprotective effects of IPC.29
Extracellular signal-regulated kinases (Erk 1/2) are members of MAP kinases, a family of serine-threonine kinases concerned with the regulation of cell proliferation, differentiation, and survival, which are activated in response to I/R, oxidative stress, hypoxia, and β-adrenergic stimulation.24 Erks also have been known to mediate cellular protection against I/R injury.30 In the present study, our results demonstrated that thiopental activates Erks and maintains cell viability (MTT assay) in neonatal cardiomyocytes exposed to hypoxia and reoxygenation. In response to an apoptotic stimulus such as hypoxia-reoxygenation, the proapoptotic protein, Bax, undergoes a conformational change that allows it to translocate to the mitochondria, inducing cytochrome c release. In addition, activaton of Erk 1/2 inhibits the conformational change in Bax protein and cytochrome c-induced caspase activation, thereby preventing apoptosis.31 The anti-apoptotic protein Bcl-2 was identified in the outer mitochondrial membrane and attenuates cellular injury by inhibiting cytochrome c translocation, preventing injurious Ca2+ release from the endoplasmic reticulum32 and inhibiting Bax translocation from the cytoplasm to the mitochondria.33 During IPC, an increase in Bcl-2 concomitant with a decrease in the proapoptotic protein Bax was observed in the isolated rat heart.34 It has been reported that overexpression of Bax can affect the storage of Ca2+ in the intracellular membranes, and changes in intracellular Ca2+ storage can feed back to the translocation of Bax from cytosol to intracellular membranes.35 Our results demonstrated that the thiopental-induced protective effect against hypoxia-reoxygenation injury was associated with an increase in Bcl-2 expression, decrease in Bax, and attenuation of mitochondrial cytochrome c release, resulting in the reduction of caspase-3 activity.
Ca2+ is one of the most important messengers in living cells for intracellular signal transduction and plays an important role in cardiac excitation-contraction coupling.36 During the process of excitation-contraction coupling, intracellular Ca2+ homeostasis is carefully regulated by ion channels, and specific binding and transport proteins. Changes in Ca2+ homeostasis play an important role in the modulation of apoptosis,35,37 and the balance of Bcl-2 and Bax on mitochondrial Ca2+ homeostasis could play a role in determining the fate of cells to survive or undergo apoptosis.37 Therefore, intracellular dysregulation of Ca2+ homeostasis is critical for cellular injury induced by hypoxia-reoxygenation. The Ca2+ entry across the membrane through L-type Ca2+ channels is balanced by the efflux of Ca2+ from the cell via the NCX and PMCA1.36 In the present results, the density of L-type Ca2+ channels and PMCA1 appeared to decrease, but the quantification of NCX was higher in hypoxia-reoxygenated cells, similar to the observations in various animal models of heart failure.38,39 In a study using mice with cardiac-specific ablation of NCX, Ca2+ entry via reverse-mode NCX is a major cause of intracellular Ca2+ overload.40 When hypoxia-reoxygenated cells were treated with thiopental, transcription levels of PMCA1 and NCX were restored to the level of normal cells, but there was no alteration in the level of L-type Ca2+ channel. It has been reported that PMCAs play a major role in maintaining a basal level of Ca2+ in brain39 and NCX is essential for maintaining Ca2+ homeostasis in cardiomyocytes.41 Our results indicate that attenuation of abnormal NCX gene expression by thiopental (p < 0.01) may be an important target for cardioprotection.
The sarcoplasmic reticulum (SR) plays a major role for regulating the intracellular Ca2+ concentration and contains SR Ca2+-cycling proteins such as RyR, SERCA2a, PLB, and calsequestrin. Reduced expression of SERCA2a and increased expression of NCX are core changes that contribute to altered Ca2+ homeostasis in the failing heart.37 Reduced expression of SERCA2a and modest, but significantly, increased expression of NCX (reverse mode) in our results would contribute to intracellular Ca2+ overload following hypoxia-reoxygenation. Treatment with thiopental restored the level of SERCA2a gene expression (p < 0.05), therefore, thiopental may play an important role in preventing Ca2+ overload by hypoxia-reoxygenation. Our results showed increased expression of calsequestrin in hypoxia-reoxygenation condition (p < 0.05), but there was no significant change in response to exposure to thiopental. Calsequestrin is located in the SR lumen and reduces ryanodine receptor activity (RyRs: Ca2+ release channels). Considering that calsequestrin overexpression increases SR Ca2+ content by inhibition of RyRs,42 calsequestrin overexpression is also likely to contribute to intracellular Ca2+ overload. Several studies have shown that expression of SR genes was increased by antioxidants and β-adrenergic receptor blockers,43,44 and further studies are required to validate the transcription mechanism regulating the expression of SR genes by thiopental.45
Mitochondria play essential roles in cell energy production, oxidative stress, and regulation of apoptosis.46 Intracellular Ca2+ overload could promote the opening of mitochondrial permeability transition pore (MPT), leading to a sudden ΔΨm collapse.47,48 During I/R, ΔΨm dissipation caused by MTP opening could lead to cell death, which in turn may affect Ca2+ homeostasis.49,50 Our results showed that thiopental alleviates hypoxia/reoxygenation-induced cardiomyocyte apoptosis through stabilizing mitochondrial ΔΨm and this is somewhat contradictory to other report that the mitoKATP channel opening was inhibited by thiopental.51 It has also been reported that MTP opening is promoted by oxidative stress.47,49,52 Therefore, mechanisms involving ROS may require further investigation. As for the limitation of our study, the experimental model used in this study does not fully mimic the complex changes in clinical conditions, and thus results using cultured myocytes should be interpreted carefully. Although we did not investigate adult rat cardiomyocytes, many studies have shown that primary cultured neonatal rat cardiomyocytes were useful models to investigate cardioprotective effects.49,52
In conclusion, hypoxia-reoxygenation stress increases intracellular Ca2+ content via enhanced NCX-mediated Ca2+ influx and decreased SERCA2a expression resulting in myocardial cell injury through intracellular Ca2+ overload in neonatal cardiomyocytes. Thiopental is likely to attenuate the abnormal changes in expression of genes related to Ca2+ homeostasis, resulting in cardioprotection through the reduction of apoptotic cell death.





 XML Download
XML Download