

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
von Hippel-Lindau (VHL) disease is an inherited autosomal dominant (AD) familial cancer syndrome, and hemangioblastomas of the cerebellum and retina, renal cell carcinoma and pheochromocytoma develop frequently in these patients. However, glioma and notably astrocytoma have been reported to occur rarely only in patients with VHL disease and in those without a history of irradiation.1-3 All previously reported cases of gliomas in patients with VHL disease were low-grade or pilocytic astrocytomas, whereas malignant glioma, especially glioblastoma multiforme (GBM), has not yet been reported in the literature worldwide. There has also never been any benign or malignant glioma due to radiation in patients with VHL disease. Our case involves a 32 year-old male with VHL disease, and he had multiple cerebellar and spinal hemangioblastomas, pancreatic cysts and a renal cell carcinoma. The largest hemangioblastoma in his right cerebellar hemisphere was irradiated with the dose of 50.4 Gy after its complete excision. It, however, recurred at the same site 7 years later and the cytomorphologic features were totally different from those of hemangioblastoma as the tumor was found to be a malignant glioma, most likely a glioblastoma multiforme. Although recurrence and invasion of cerebellar hemangioblastomas have generally been reported as their clinical behavior,4,5 either malignant transformation of the hemangioblastoma or radiation-induced malignant glioma in a patient with VHL disease has not yet been reported in the literature worldwide. The most common genetic alteration in VHL disease is known to be allelic loss at chromosome 3p25-26. Most cases of radiation-induced astrocytomas have been reported to have p53 mutation.6 We report herein the first case of malignant glioma in a patient with VHL disease, and this tumor developed in the previous excision site of a hemangioblastoma after postoperative irradiation. We also tried to find the genetic origin of the malignant glioma by examining the genetic alterations in both the hemangioblastoma and the malignant recurrent tumor.
CASE REPORT
Clinical summary
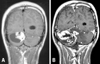
A 25-year-old male first visited our hospital in 1998 with the chief complaints of headache and dizziness of a remote onset. He had experienced the symptoms for about 4 years, but they were aggravated 4 - 5 months before the hospital visit and they were accompanied with pain on both eyeballs and bilateral intermittent tinnitus. Magnetic resonance imaging (MRI) of the brain and cervical spine revealed a 4×4.5×6 cm-sized cystic tumor with a 2×3 cm-sized enhancing mural nodule at the right cerebellar hemisphere (Fig. 1A). The cystic tumor had a heterogeneous rim and accompanying hydrocephalus. There were also multiple small enhancing nodules in the left cerebellar hemisphere and cervical spinal cord. The pancreas and kidney showed multiple cysts and bilateral cortical simple cysts, respectively. He was diagnosed as having von Hippel-Lindau disease, based on all these radiographic findings. Under the impression of hemangioblastoma, he underwent a complete excision of the right cerebellar tumor. One month after excision, he received a conventional radiotherapy using 6 megavolume (MV) of photon on both cerebellar hemispheres and cervical spinal cords, with a total dose of 50.4 Gy being divided by 28 times for 47 days. The postoperative radiotherapy was performed to treat both the operated and non-operated multiple hemangioblastomas. The patient made an uncomplicated recovery for the excised hemangioblastoma and underwent an uneventful clinical follow-up for more than 7 years.
At the age of 32 years in 2005, however, he experienced headache and dizziness again for about 5 weeks before his latest visit to the hospital. Brain MRI revealed a big right cerebellar cystic mass with a total size of 5.8×3.7×3.1 cm. The mass showed heterogeneous and irregular-shaped enhancement along the thickened walls and the big non-enhancing necrotic center (Fig. 1B). It was radiologically suspicious of malignant change, because of the increased size compared to the previous one, the irregular and heterogeneous enhancement and the marked peritumoral edema extending to the contralateral cerebellar hemisphere. The right kidney showed a 1.9×1.5×1.3 cm-sized low-attenuated mass at the upper pole. He underwent two operations of the right cerebellar mass removal and right nephrectomy, within an interval of about 2 weeks. The cerebellar mass, however, was growing very rapidly right after the second removal to form a big cystic and solid tumor again with the size of 7.5×5.8×5.0 cm after about 40 days. Brain MRI revealed increased size and extent of invasion, with diffuse enhancement and peritumoral edema. The third removal of the cerebellar tumor revealed the same pathologic findings as the previous recurrent tumor. When he was 33 years-old in 2006, the right cerebellar tumor showed two more recurrences at the same site. He once more underwent excision of the tumor and then he died after last tumor recurrence.
Pathologic findings
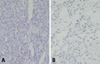
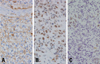
The excised mural nodule of his first right cerebellar tumor revealed a homogeneously rich cellularity and vascularity with the main two tumor cell components being stromal cells and fine capillary networks. The stromal tumor cells were round to polygonal with foamy abundant cytoplasm, showing a diffuse immunopositivity for S100 protein (Fig. 2A). The intervening capillaries showed a fine intricate netlike arrangement, which was delineated better in the CD34 immunostaining (Fig. 2B). Both p53 and glial fibrillary acidic protein (GFAP) immuno-stainings were negative in the stromal tumor cells (Fig. 3). All these findings were compatible with hemangioblastoma.
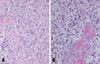
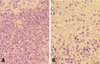
The recurrent cerebellar tumors had cellularity even higher than that of the first one, with compactly arranged polymorphic tumor cell components in the fibrillary stromal background. The tumor cells showed largely short-spindled to polygonal shapes with eosinophilic granular or fibrillary cytoplasms, and with marked nuclear atypia and frequent mitoses, which were compatible with malignant glioma (Fig. 4A). Many polygonal cells with clear vacuolated cytoplasms were aggregated in large sheets, and they looked like oligodendroglial cells (Fig. 4B). Some areas also showed small to medium-sized anaplastic tumor cells with high nucleocytoplasmic ratio and frequent mitoses (Fig. 5A). Prominent endothelial proliferation, spotty necrosis and occasional tumor giant cells were noted (Fig. 5B). Based on all these microscopic findings, these pleomorphic anaplastic tumor cells could be diagnosed as a malignant glioma, most likely GBM. Immunohistochemical study revealed diffuse GFAP positivity in the tumor cells (Fig. 6A). The tumor cells were also highly positive for p53 immunostaining (Fig. 6B), which is indicative of p53 mutation. In addition, there was loss of p16 immunoreactivity (Fig. 6C), although either epidermal growth factor receptor (EGFR) overexpression or deletion of retinoblastoma (RB) gene was not noted. The tumor was finally diagnosed as malignant glioma, and it was most likely a glioblastoma multiforme of astrocytic origin. There was no evidence of any residual hemangioblastoma in the recurrent tumors.
Genetic study
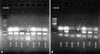
We extracted DNA from the cerebellar hemangioblastoma, recurrent malignant gliomas, and renal cell carcinoma from their paraffin blocks, along each normal tissue counterpart within the same block. We performed a chromosomal DNA polymerase chain reaction (PCR) to compare and evaluate the qualitative change such as an allelic loss among the tumor and normal groups. Loss of heterozygosity (LOH) of the initial and recurrent tumors was analyzed with three microsatellite probes (D3S 1038, D3S 1317, and D3S 1304) for 3p and one polymorphism marker (A1149G) for the VHL gene.7 Only the cerebellar hemangioblastoma showed allelic losses for two genetic loci of 3p25-26 (D3S 1038 and D3S 1304) (Fig. 7). The allelic loss at the genetic locus detected by D3S 1304 microsatellite probe was more severe than that by D3S 1038 probe. The malignant glial tumor and renal cell carcinoma revealed no evidence of LOH for all four oligonucleotide primers. The results of the genetic and immunohistochemical studies are summarized in Table 1.
DISCUSSION
Astrocytomas arising in VHL disease have been reported mostly as pilocytic and low-grade cerebellar astrocytomas, which developed as either primary or recurrent tumors in patients with VHL disease.1-3 The reported cases had no history of radiation therapy. However, any malignant glial neoplasms in VHL disease either by radiation or not have not yet been reported in the literature worldwide. The present case displayed a recurrent cerebellar tumor 7 years after his right cerebellar mass had been excised and then irradiated with 50.4 Gy of high dose radiation. The recurrent tumor showed totally different histopathology in comparison to the hemangioblastoma, and the recurrent tumor had a history of high-dose radiation therapy, diffuse GFAP immunopositivity, p53 mutation and no LOH for the VHL gene. The recurrent tumor was, therefore, considered to be a radiation-induced malignant glioma rather than malignant transformation of the hemangioblastoma, and it fulfilled the criteria for the radiation-induced neoplasm. The criteria for radiation-induced neoplasm are generally known to be as follows;8-10 1) the tumor must appear in the irradiated area, 2) the tumor must not be present prior to irradiation, 3) a sufficient latency period must elapse between irradiation and the appearance of the tumor, 4) the radiation-induced tumor must be histologically proven and it must be of a different histological type from the original first tumor that was treated by radiation therapy, 5) the incidence of second lesions is significantly higher than that in controls, and 6) there must be no known genetic or other predisposing conditions for tumor development. The present case fully met the criteria 1 through 5, but not 6 because he has a genetic basis for VHL disease, which is known to be an AD tumor-prone syndrome. However, VHL disease patients have not yet been reported to develop any benign or malignant tumors after radiation therapy.11 Therefore, it is considered that the 6th criteria can be applied loosely to the radiation-induced tumors in the VHL disease patients, because they don't seem to be genetically vulnerable to the radiation. On the other hand, other AD tumorprone conditions such as neurofibromatoses types 1 and 2 have frequently been reported to have benign or malignant neoplasms like malignant peripheral nerve sheath tumor (MPNST) that occurred after radiotherapy.11 It would, therefore, appear that patients with genetic disorders are particularly sensitive to radiation carcinogenesis due to their relative genetic instability.11 Malignant glioma spontaneously arising in a cerebellar hemangioblastoma has not yet been reported either. Our case didn't seem to show malignant transformation from the cerebellar hemangioblastoma, as the hemangioblastoma had no GFAP immunopositivity and it was grossly completely excised, with no residual tumor after the first operation and before irradiation.
The carcinogenesis of radiation-induced malignant gliomas is not clear, but ionizing radiation is well-known to induce genomic instability, which is transmitted over many generations through the progeny of the surviving cells. Since the induced genomic instability accumulates gene mutations and gross chromosomal rearrangements, it has been thought to play a role in radiation-induced carcinogenesis. It has also been suggested that radiation-induced neoplasms are caused by imperfect repair of delayed ionizing radiation-induced DNA strand breaks in the tumor suppressor genes or the protooncogenes.6,12,13 Radiation-induced gliomas are generally known to be high-grade at clinical presentation and they are mostly astrocytic in their differentiation. Our case showed malignant glioma with diffuse GFAP immunopositivity, therefore, could be diagnosed as GBM of an astrocytic origin. These tumors have also been reported to have genetic alterations similar to primary spontaneous high-grade astrocytoma.6 They include EGFR amplification, p16 deletion and p53 mutation.6 In the present case, immunohistochemical staining showed p53 mutation and p16 deletion in the malignant glioma, although either EGFR overexpression or RB deletion was not detected. These genetic alterations of the malignant glioma suggest that it occurred as a result of radiation-induced genomic instability of the surviving cerebellar glial cells without the genetic alteration of VHL disease.
The primary tumors which are known to develop postirradiation malignant gliomas have been reported to be pituitary adenoma, medulloblastoma, retinoblastoma, malignant lymphoma, acute leukemia, thyroid carcinoma and meningioma.8,10,14-16 Cerebellar hemangioblastoma has never been listed to date in the literature as a primary tumor that can evoke postirradiation malignancy. However, we experienced a case of cerebellar hemangioblastoma in a patient with VHL disease, and the patient developed malignant glioma after receiving 50.4 Gy of irradiation at the original complete resection site. This case report stresses the fact that radiation as an adjuvant therapy of hemangioblastoma should be used and monitored more strictly in a patient with VHL disease because a malignant glioma such as GBM can develop several years later at the irradiation site.


 XML Download
XML Download