

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Aberrant promoter hypermethylation or histone modifications are recognized as an important mechanism in cancer initiation and progression.1 These epigenetic mechanisms are recognized as a "third pathway" in Knudson's model of tumor-suppressor gene inactivation and can affect gene expression without accompanying genetic changes.2 DNA methylation occurs in the cytosine-guanine sequence (CpG) in mammalian DNA strands. The pattern of methylation at cytosine residues in the CpG sequences is established during early development and is heritable.3 In humans, approximately 70% of CpG dinucleotides are methylated. The frequency of the CpG dinucleotide is relatively low in most of the human genome because of CG suppression. However, genomic sequencing reveals that CpG sites are unevenly distributed in the mammalian genome. About 60% of genes have 5' promoter regions located in the CpG rich 0.3-2 kb stretches of DNA called CpG islands. CpG islands are normally unmethylated.4,5 Promoter associated CpG islands need to be protected against DNA hypermethylation that results in irreversible inhibition of gene expression. In normal cells, this is evidenced by methylation associated silencing of imprinted genes and inactive X chromosome genes. In cancers, this mechanism is utilized to inactivate tumor-suppressor genes.6
Histone modification is another key mechanism in transcriptional regulation, which is well conserved through a variety of species.7-9 The less structured N-terminal domains of all core histones protrude from the core histone and are subject to chemical modifications, such as acetylation, methylation, and phosphorylation at certain residues.10,11 The modification patterns of histone have been linked to biological function and act as a "histone code", which implies that transcription states can be predicted by simply deciphering this code.8 Recent studies showed that histone modifications co-operate with DNA methylation to affect gene inactivation in some tumor suppressor genes.12-16 DNA methylation and histone modifications might interact with each other and establish stable gene silencing. In addition to this interaction, histone H3 lysine 27 trimethylation (H3K27me3), which is involved in the regulation of homeotic (Hox) gene expression and in the early steps of X-chromosome inactivation in women, can be solid silencing machinery that is independent of DNA methylation in human cancers.17-19
Although evidence for aberrant DNA methylation and histone modification changes in cancers is accumulating, how aberrant DNA methylation and histone modifications interact in a certain loci, why some genes are particular targets of epigenetic machinery, and whether aberrant DNA methylation and histone modifications confer specific roles in cancer initiation and progression remains unclear.
In this review, we discuss the current understanding of gene silencing by DNA methylation and histone modifications, focusing especially on histone H3 lysine 9 methylation (H3K9me) and H3K27me3, as related to human cancers.
Histone modifications
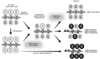
Nucleosomes consist of 147 bp DNA wrapped around the core histone that comprises histone proteins, H2A, H2B, H3, and H4 (Fig. 1A).7,20 The N-terminal domains of all core histones are subject to chemical modifications, such as acetylation, methylation, ubiquitylation, and phosphorylation at certain residues.8 Histone modifying enzymes bring the complexity of post-translational modifications that can either activate or repress transcription, depending on the type of chemical modification and its location in the histone protein. The modification pattern of histone has been linked to chromatin structure and gene function during development as well as tumorigenesis.6,10,11
These different combinations of histone tail modifications influence transcription by affecting chromatin structures.8 Modifications on the lateral surface of the core histone could also affect histone-DNA interactions as well as N-terminal domains. The respective enzymes vary in their potential to induce mono-, di-, or tri-methylation (Fig. 1B). Since lysine methylation is more stable than other modifications, there has been until recently, discussion as to why stably methylated histone lysine residues contributed to the establishment and propagation of different patterns of gene expression in the same genome.21 Recent comprehensive reports revealed that histone methylation is reversible by demethylases (Fig. 1B). Given that histone methylation has important roles in development and other biological events, including tumorigenesis, histone demethylases might be key molecules in cellular processes.
Generally, acetylation of lysine residues on histone H3 and H4 leads to the formation of an open chromatin structure, which indicates transcriptional activity.22 In contrast, methylation on lysine residues link to either activation or repression depending on its location.11 Trimethylation at K9 on histone H3 (H3K9me3) or K20 on histone H4 (H4K20me3) has been shown to form heterochromatin from yeast to humans. Dimethylation at K9 (H3K9me2) is associated with inactivation of gene expression at the euchromatic region. Trimethylation at histone H3K27 (H3K27me3) is a distinct histone modification involved in the regulation of homeotic (Hox) gene expression and in the early steps of X-chromosome inactivation in women.23 This process is mechanistically linked to polycomb group (PcG) proteins. PcG proteins, an enhancer of zeste 2 (EZH2), which is a member of polycomb repressor complex 2 (PRC2), has histone methyltransferase activity with substrate specificity for H3K27. H3K27me3 serves as a signal for specific binding of the chromodomain of another polycomb repressor complex, PRC1, which includes B lymphoma Mo-MLV insertion region 1 (BMI-1), ring finger protein 1 (RING1), human polycomb (HPC), and human polyhomeotic homologue (HPH).24 The binding of PRC1 blocks the recruitment of transcriptional activation factors, such as SWI/SNF, and the presence of PRC1 prevents initiation of transcription by RNA polymerase II.25,26 Di- or tri-methylation on K4 on histone H3 (H3K4me2/me3) localizes to sites of active transcription and this modification may be stimulatory for transcription. Several recent studies indicated the existence of 'bivalent' domains consisting of dual marks of repressive histone H3K27me3 and the activation of histone H3K4me3 modifications on development-associated genes that are silent in ES cells and activated during differentiation (Fig. 2).27,28 The dynamic alteration of lysine methylation contributes to reversible and plastic regulation of gene expression in varieties of the cellular process, which contrasts with stable gene inactivation by DNA methylation.
DNA methylation in cancer
Promoter DNA methylation is the most widely studied epigenetic modification of human cancers.29 Compared to normal tissues, cancer DNA shows global hypomethylation and hypermethylation at gene-associated CpG islands that are normally unmethylated. Cancer hypomethylation is found in repetitive elements localized in satellite sequences or pericentromeric regions and has been reported to lead to genomic instability.30 Hypermethylation in CpG islands is found in several types of cancers and recognized as a mechanism of tumor suppressor gene silencing. Initial studies indicated DNA methylation as a cause of tumorigenesis. For example, inactivation of the cyclin-dependent kinase inhibitor P16/CDKN2A by methylation leads to the disruption of cell-cycle regulation and potentially provides a growth advantage to affected cells.31 Another tumor suppressor genes, P14/ARF is also a target of inactivation by DNA methylation.32 P14/ARF activates P53 through interactions with MDM2, and losses of P14/ARF negatively affect P53 function. Inactivation by methylation has also been found in DNA repair genes, such as hMLH1 and MGMT.33,34 Tumor suppressor gene silencing by DNA methylation promotes cell proliferation and may provide strong selective advantages. However, potential oncogenes, such as COX2 and hTERT, are also targets of aberrant DNA methylation, which illustrates the point that methylation is initially independent of gene function.35,36 These data suggest that cancer-associated DNA methylation is not restricted to tumor suppressor genes and that the profile of methylation in cancers may not simply be a result of selection.
Recent accumulating evidence suggests that DNA methylation is a late step in the gene silencing process, even though such methylation has been detected in pre-cancerous tissues. The establishment of fundamental DNA methylation patterns during early development might be accomplished through histone modification.37 H3K4me has recently been suggested to protect gene promoters from de novo DNA methylation in somatic cells.38,39 Therefore, de novo methylation is closely associated with the removal of methylation from H3K4 by Lsd1 and jmj-C containing demethylases [Jarid 1a and Jarid 1b, also known as K (lysine)-demethylases 5A (KDM5A) and K (lysine)-demethylases 5B (KDM5B)] on certain loci.40-42 After removal of methylation from H3K4, Dnmt3A/Dnmt3B-Dnmt3L complexes can target these loci, resulting in de novo methylation.37,43 However, H3K4me marks most CpG islands, since RNA polymerase II recruits specific H3K4 methyltransferases, which link to CpG islands of actively transcribed genes.39,44 Contact between DNMT3L and nucleosomes are inhibited by H3K4me. As a result, most CpG is prevented from de novo methylation.
The DNA methylation pattern is maintained by DNA methyltransferase 1 (Dnmt1), which is associated with a replication complex. Studies have indicated that Dnmt1, together with UHRF1 (also known as Np95 or ICBP90), recognizes the hemimethylated CpG residues and methylates the opposite strand, resulting in a faithful copy of the methylation profile of the parent cell.45 Regulation of Dnmt1 methylation by histone demethylase Lsd1 and histone methylase Set7/9 has recently been reported.46 According to this model, Lsd1counteracts methylation of Dnmt1 by Set7/9, which results in the stabilization of Dnmt1 and enables DNA methylation to be maintained.
Once DNA methylation has been established on CpG islands, this modification is generally irreversible without artificially altering key factors in the cells, as was shown in iPS cells.47,48 Therefore, facultative regulation by histone modifications is stably determined by DNA methylation on the silenced loci in cancer cells.
Interrelation between DNA methylation and histone modifications
Early studies have shown that the link between DNA methylation and histone modifications is mediated by a group of proteins with methyl DNA binding activity, including methyl CpG binding protein 2 (MeCP2), Methyl-CpG binding domain protein 1 (MBD1), and Kaiso [also known as ZBTB 33 (Zinc finger and BTB domaincontaining protein 33)]. These proteins localize to DNA methylated promoters and recruit a protein complex that contains histone deacetylases (HDACs) and histone methyltransferases.49-51 These studies suggest DNA methylation induce chromatin structural changes through alteration of histone modifications. It is known that DNA methylation inhibits H3K4me, which is also evidence that DNA methylation affects histone modifications.38,39 However, early studies in fungi (Neurospora crassa) show that mutations of histone H3K9 methyltransferase reduced DNA methylation, indicating a simple linear model in which H3K9 methylation acts as an upstream epigenetic mark that signals to DNA methylation.52 In embryonic stem cells, Oct3/4 is inactivated after lineage commitment. For this silencing process, a repressor complex is recruited that contains histone methyltransferase G9a and enzymes with histone deacetylase activity. Subsequently, methyltransferase, DNMT3A, and DNMT3b, which catalyze de novo DNA methylation, are recruited at the promoter.13 Intriguingly, interaction between G9a and the DNMTs depends on the ankyrin motif of G9a.53 By contrast, the SET domain, which is responsible for the methyltransferase activity of G9a, does not interact with DNMTs. Indeed, mutation of the SET domain is inert in methyltransferase activity and disrupts H3K9 methylation without affecting DNA methylation.54,55 These data suggest that DNA methylation on the promoter depends on the recruitment of G9a (especially ankyrin motif), rather than the histone methyltransferase activity itself. The interactions between DNA methylation and histone H3K9 methylation currently fit a model whereby these two changes form a reinforcing silencing loop or bidirectional crosstalk, and this may explain why silencing is less stable in organisms that lack DNA methylation (Fig. 3).2
Recently, links between PcG-mediated methylation on H3K27 and de novo DNA methylation in cancers were described using ChIP analyses coupled with bioinformatic database mining, which supports the conventional view that the mark imposed by PRC2 during development may predispose some genes for later de novo methylation (Fig. 3).14-16 Genome-wide analysis combined with methylation prediction models revealed that CpG islands predicted to be methylation-prone revealed a strong association with embryonic targets of PRC2 and a subset of PRC2 targets that were more likely to be hypermethylated in cancer.56 Biochemical study also showed that DNMTs bind to EZH2, however, this interaction seems to be detected only under certain conditions.48,57,58 Obviously, the presence of histone methylation at H3K9 or H3K27 does not always lead to de novo methylation. Indeed, subsets of PcG target genes are methylated in one cancer, even though these same genes are completely unmethylated in another cancer from the same normal tissue origins. Additional factors are required for triggering DNA methylation in the genes enriched with those histone modifications.
More recently, comprehensive genome-wide and functional analysis revealed that PcG-mediated H3K27me3 is a mechanism of tumor-suppressor gene silencing in cancer that is potentially independent of promoter DNA methylation and a direct interaction between PcG-mediated methylation on H3K27 and de novo DNA methylation pathways is unlikely in cancers.18,19 This independence of DNA-methylation and histone marking appear to conflict with previous studies. It must be noted that the majority of genes enriched with repressive mark H3K27me3 in prostate cancers do not have the CpG island in the promoter regions, whereas genes targeted by PcG complexes are generally associated with CpG island promoters in ES cells.18,48 This indicates that targets of PcG-mediated methylation on H3K27 among ES cells, normal tissue cells, and cancer cells are different and that cancer cells usurp the silencing mechanisms to extinguish functional pathways by H3K27me3. Now, three lines might be considered in the silencing machinery associated with PcG-mediated methylation on H3K27 (Fig. 4). First, genes are de novo repressed by PcG-mediated methylation on H3K27, which supports the observation of distinct target genes between normal and cancer cells (particularly targeting non-CpG island promoters). Second, during early tumorigenesis, subsets of genes become methylated, and a large portion of these are initially marked by the PcG complex (targeting CpG island promoters). In this case, several genes undergo epigenetic programming, in which genes that are originally silenced by PcG acquire DNA methylation as an alternative silencing mechanism. This epigenetic switch to DNA methylation-mediated repression reduces the epigenetic plasticity, locking the silencing of key regulators and contributing to tumorigenesis.19 Third, genes that are originally silenced by PcG acquire DNA methylation to some extent. In this case, H3K27me3 and DNA methylation co-exist on the same promoter and PcG-mediated H3K27me3 is the dominant silencing machinery, which can be reactivated by inhibition of PRC complexes. In cancers it appears that different forms of gene silencing contribute to tumorigenesis, which range from a flexible and plastic repressor-based mechanism to a highly stable inactivation that is maintained by DNA methylation.
Future direction: perspectives on epigenetic therapy
This review has focused on the relationship between two major epigenetic mechanisms: DNA methylation and histone methylation. Clarifying this relationship is important in the context of cancer epigenetic therapy. Restoring gene function silenced by epigenetic changes in cancer has the potential of 'normalizing' cancer cells by epigenetic therapy. This could have a serious impact on the prevention and treatment of human cancers. However, there are important questions that must be understood in regard to human cancers and how DNA methylation affects chromatin structure and vice versa remain. It will be important to elucidate the precise role of these epigenetic mechanisms that allow cancer cells to revert to a more normal state through epigenetic reprogramming.
HDAC inhibitors lead to the accumulation of acetylation in histones, which results in reversion of chromatin status and transcriptional activity to a normal state.59 HDAC inhibitors have been shown to induce P21, which is responsible for cell cycle arrest and cell differentiation. Another possible role of HDAC inhibitors in antitumor activity might be to inhibit PcG proteins, some of which have been recognized as important and uniquely acting oncogenic proteins, in cancers. Since PcG-mediated gene silencing is initiated by the HDAC activity of PRC2, inhibition of HDAC can efficiently reactivate the H3K27me3 target genes.18,60
DNA methylation is a therapeutic target of human cancers.59,61 The cytosine analogues, 5-azacytidine and 5-aza-2'-deoxycytidine, are powerful inhibitors of DNA methylation, which are incorporated into DNA during cell division. They trap DNA methyltransferases and lead to cell differentiation and growth repression. These demethylating agents have received FDA approval for the treatment of myelodysplastic syndrome. However, transiently induced demethylation is impaired after the withdrawal of chemicals, which has been suggested in cell line studies.62 This phenomenon might be partially explained by epigenetic memory retained in chromatin, which may include histone methylation and interfere with the complete epigenetic reprogramming in cancer cells, as shown in this review. Combining DNA methyltransferase and histone methyltransferase inhibitors might provide a clue to solving this incomplete reprogramming. In addition, the synergistic effects between DNA methyltransferase and HDAC inhibitors suggest clinical trials of this approach to restore gene function silenced by aberrant chromatin changes in cancers.63 Nevertheless, complete understanding of these epigenetic modifications and their cross-talk will lead to the development of the most effective therapies possible. With such knowledge of epigenetics at our disposal, we may yield the full capacity to not only treat but to prevent cancer. By correcting epigenetic abnormalities that predispose to cancers, the promise for epigenetic therapies as an essential treatment option will be achieved.


 XML Download
XML Download