

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Plasma ultrafiltration during a primary urine formation in the glomerulus is a central function of the kidneys. This process is associated with the anatomical region of the kidney, which is composed of a basement membrane with highly specialized epithelial cells called podocytes.1 As a part of the glomerular filtration barrier, the slict diagram (SD) is thought to function as a filter that is size-selective. In the nephrotic syndrome, the normal podocyte substructure is lost; the effacement of podocyte foot processes results in a massive proteinuria.2,3 During the development of the nephrotic syndrome, this elaborate morphology of the podocyte is dramatically altered, resulting in changes such as cellswelling, retraction, spreading of the foot processes, vacuole formation, formation of occluding junctions with displacement or disruption of the slit diaphragms, and foot process detachment from the glomerular basement membrane.4-8 The cellular and molecular changes that occur within the podocytes have been identified by the detection of mutations in several genes encoding podocyte proteins, including CD2AP, nephrin and podocin.9-11 Here, we selected podocin, a component of the glomerular filtration barrier, as a target for the yeast 2-hybrid system.
Podocin is an integral membrane protein with a single membrane domain that forms a hairpin-like structure which places both the N- and C-termini in the cytosol. As a membrane protein, it is almost exclusively expressed in a subset of highly specialized kidney epithelial cells, called podocytes, localized at the insertion site of the SD complex in the podocyte foot processes. Furthermore, it is a constituent of detergent-insoluble microdomains and has been shown to interact with another raft-associated podocyte proteins, CD2-associated protein (CD2AP) and nephrin.12 Hence, podocin may act as a scaffolding protein in the podocyte lipid rafts, recruiting nephrin and CD2AP to these microdomains. Podocin has been shown to play a major role in glomerular permeability; mutations in this gene result in early onset nephrotic syndrome. Therefore, we screened a mouse kidney complementary DNA (cDNA) library to identify proteins that interact with podocin in nephrotic syndrome.
MATERIALS AND METHODS
Amplification of the podocin genomic sequence from the mouse kidney
A frozen mouse kidney tissue sample (100 mg) was homogenized in TRIZOL reagent (Gibco, Carlsbad, CA, USA), and total RNA was then isolated by addition of a mixture containing chloroform, isopropyl alcohol and 75% ethanol solution. Using the Omniscript Reverse Transcription kit (Qiagen, Hilden, Germany), cDNA was synthesized with total RNA (1 µg) as the starting template. To amplify the podocin gene from the mouse kidney cDNA, podocin primers were designed (EcoR I-containing forward primer 5'-GGAATTCATGGACAGCAGG-3' and BamH I-containing reverse primer 5'-CGGGATCCCTAACATAGGAGA-3') based on a homology search, and cDNA and primers were added to the Polymerase Chain Reaction (PCR) PreMix kit (iNtRon, Seongnam, Korea). PCR conditions were as follows: 1 cycle of 95℃ for 3 minutes, 30 cycles of 95℃ for 30 seconds, 57℃ for 30 seconds, and 72℃ for 80 seconds followed by one cycle of 72℃ for 7 minutes. The PCR product was run on a 1% agarose gel containing ethidium bromide, and then visualized under ultraviolet (UV) light.
Cloning and sequencing of the mouse podocin
Amplified PCR products eluted from agarose gel were obtained using PCRquick-spin™ (iNtRon). The purified PCR product and the pGBKT7 vector were cut with the restriction enzymes, EcoR I and BamH I (Roche, Mannheim, Germany), at 37℃ for 1 h, respectively. They were ligated using T4 DNA Ligase (Roche) for 16 h at 16℃. The resulting construct, pGBKT7 + podocin, was transformed to an ECOS competent cell (DH5α) by heat shock as described previously.13 Some colonies selected from the plate were cultured in 5 mL of Luria broth (LB) with 100 µg/mL ampicillin for 16 hours at 37℃ and extracted using the Plasmid DNA-spin™ purification kit (iNtRon). After adding the restriction enzymes to the eluted plasmid, automated DNA sequencing was performed to identify plasmids with the podocin gene. The BLASTN algorithm was applied to search the National Center for Biotechnology Information (NCBI) non-redundant nucleotide database.
Yeast transformation
Using the lithium acetate (LiAc)-mediated yeast transformation protocol (Clontech, Palo Alto, CA, USA), the bait plasmid, pGBKT7-podocin, was transformed into the yeast strain AH109, and then spread out on a SD/-Trp/-Ura plate. The colonies that appeared on the SD/-Trp/-Ura plate were cultured in SD/-Trp/-Ura media and confirmed to produce AH109/pGBKT7-podocin by colony PCR. The mouse kidney cDNA library (normal, whole kidneys pooled from 200 BALB/c, ages 8-12 weeks) was then transformed into the AH109/pGBKT7-podocin, and the amplified yeast co-transformants were plated on a SD/-Trp/-Leu plate (low stringency). To screen for positive clones that interact with podocin, the colonies plated at high density on the SD/-Trp/-Leu plate were scraped off and then transferred to the SD/-Trp/-Leu/-His plate (medium stringency). Then, the colonies that appeared on the SD/-Trp/-Leu/-His plate were transferred to the SD/-Trp/-Leu/-His/Xα-Gal plate.
Analysis and verification of the putative clones
The positive clones that were blue on the SD/-Trp/-Leu/-His/X-α-Gal plate were lysed in 200 µL of lysis buffer [0.1 M Tris-HCl (pH 8.0), 50 mM EDTA, 1% SDS] and 200 µL of phenol : chloroform : isoamyl alcohol, and vortexed at the highest speed for 5 min. The debris was removed by centrifugation at 14,000 rpm for 10 minutes. The aqueous (upper) phase was transferred to a fresh tube, and 3M acetate and 100% ethanol were added. The sample was incubated at -70℃ or in a dry-ice/ethanol bath for 1 h and centrifuged at 14,000 rpm for 10 minutes. The pellets were washed with 1 mL of 70% ethanol and dried at room temperature. The pellet was resuspended in 20 µL of H2O. The positive plasmids were transformed into Escherichia coil (E. coil), by heat shock. To select for transformants containing only the AD/library plasmid, the yeast-purified plasmids were plated on LB medium containing ampicillin. The plasmids, containing the pACT2-specific inserts purified from the Escherichia caoli (E. coil), were digested with Hind III and analyzed by agarose/EtBr gel electrophoresis. After the inserts with variant patterns were selected, they were sequenced and then the sequences were entered into the program at the website http://www.ncbi.nlm.nih.gov/BLAST to perform a BLAST analysis.
RESULTS
Amplification of the podocin gene and cloning
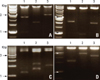
Using the primers with mouse nephrosis 2 homology, NPHS 2, the podocin gene was successfully amplified to obtain the 1,158 bp predicted fragment by PCR. Purified PCR product and pGBKT7 were ligated and transformed into the E. coli strain DH5α by heat shock. By adding the restriction enzymes, EcoR I and BamH I, the resulting plasmids with the target gene were divided into 2 bands, the insert podocin and the vector pGBKT7 (Fig. 1). These products were sequenced and confirmed to be the podocin gene by the BLASTN algorithm.
Yeast 2-hybrid screening
The podocin bait (called pGBKT7-podocin) used in the yeast 2-hybrid system contained the full-length cDNA fused in-frame with the GAL4 DNA-binding domain of pGBKT7. The pGBKT7-podocin was first transformed into AH109 by the lithium acetate-mediated method. To confirm the transformation, colony PCR was performed with a podocin primer, and confirmed to be the size of podocin on the agarose gel (Fig. 2). To screen proteins interacting with podocin, the mouse kidney cDNA library which contained the GAL4 activation domain fused into random cDNA was transformed into the AH109/pGBKT7-podocin and spread onto the SD/-Trp/-Leu plate (low stringency). After the incubation of the plates at 30℃ until colonies appeared, there were several clones at high density on the SD/-Trp/-Leu plate (Fig. 3A). This environment provided an initial phase of growth that maximized the plasmid copy number, which resulted in higher levels of the fusion protein. Clones on the SD/-Trp/-Leu plate represented weak or transient interaction of the cDNA library (AD/library plasmid) with the AH109/pGBKT7-podocin. To screen for the actual putative proteins, the clones on the SD/-Trp/-Leu plate were scraped off and then transferred to the SD/-Trp/-Leu/-His plate (medium stringency). After this step, at least 1.5-3 times the number of independent clones in the library was screened (Fig. 3B). Subsequently, colonies on the SD/-Trp/-Leu/-His plate were transferred to the SD/-Trp/-Leu/-His/X-α-gal plate (Fig. 4). As a final step to identify the AD fusion proteins that interact with podocin, all false positive interactions were virtually eliminated by this screen, however, the low-affinity protein interactions may have been missed. Clones that were blue on the SD/-Trp/-Leu/-His/X-α-gal plate were thought to be the result of AD/library plasmids mated with the plasmids encoding the AH109/pGBKT7-podocin (Fig. 4A). To identify them, they were cultured in SD/-Trp/-Leu/-His media and isolated by following the instructions in the manual (see the methods section). The plasmids isolated from the positive clones were transformed into E. coli by heat shock. After eluted from the purification kit, the plasmids digested with Hind III demonstrated different patterns on the agarose/EtBr gel (Fig. 5).
Sequencing of putative positive clones and BLAST analysis
The plasmids with the variant patterns were sequenced and analyzed using the BLAST software available on the internet. The data of the sequencing results are reported in Table 1. We identified 12 clones that were plasmids which were screened by the yeast 2-hybrid system and only 4 of the 12 clones were found to be associated with the podocyte. The biology of the 4 clones identified as plasmids that interact with podocin will require a further study.
DISCUSSION
The SD is primarily responsible for size selectivity of the glomerular filter.14,15 Previous genetic studies revealed that nephrin, CD2AP and podocin are required for normal filtration function of the SD.10,16-19 They are associated with the lipid rafts of the SD, and podocin not only co-localizes with but also interacts with CD2AP and nephrin. Podocin plays a major role in glomerular permeability; it's mutations result in early onset nephrotic syndrome in humans.9,20 A number of mutations of the podocin gene have been shown to cause the autosomal recessive, steroid-resistant nephrotic syndrome, characterized by resistance to steroid therapy and rapid progression to endstage renal disease, resulting in the need for organ transplantation.21,22 This suggests a regulatory role for podocin in determining glomerular permeability.
Here, we identified proteins that interact with podocin using the yeast 2-hybrid system. The podocin gene of the mouse kidney was amplified by PCR and isolated by cloning. The plasmid was transformed into the AH 109, yeast strain, where the mouse kidney cDNA library was then transformed. The AH 109 transformants were screened via high-, medium- and low-stringency steps. These selection steps provided less stringent screens that increased the number of false positives and the more stringent screens that decreased the likelihood of false negatives. Because the high stringency media provided a step to eliminate false positive interactions, the resulting blue clones screened by a final step were predicted to be candidate proteins for interaction with podocin. Among the clones identified by sequencing, we selected only 4 putative proteins by searching for a relationship between podocin and candidates; they were kinesin, glutathione peroxidase 3 (Gpx3), sterol carrier protein 2 (Scp2) and cathepsin H (Ctsh).
However, nephrin, CD2AP and some other podocyte-specific proteins that associate with podocin were not screened through the yeast 2-hybrid system. As a tool of the genomic approach, the yeast 2-hybrid system indicates which protein encoded in the genome of interest is expressed for the examination of mutual interactions. Furthermore, it detects not only transient and unstable interactions, but also predicts possible interactions. Therefore, it is currently so well-established to be used in a genomewide scale.23 On the other hand, this system has various limitations inherent to its method itself. According to 2 studies,24,25 the data obtained by this system did not largely overlap with each other, and failed to recapitulate the 2-hybrid interactions that are identified in conventional studies. In experiments that identify a protein-protein interaction, the percentage of their false positive and false negative is 30-60 and 40-80, respectively.24,25 The reasons for this small coverage are insufficient depth of the screening, potential misfolding or mutations interfering interactions and the use of full-length proteins.26,27 Therefore, we exists a possibility that nephrin, CD2AP and some other podocyte-specific proteins were not detected by this approach because of these reasons.
First, kinesin among the 4 putative proteins is a protein related to the podocyte process. Podocyte formation is mechanically dependent on microtubules (MT) that are regulated by various microtubule-associated proteins (MAPs). The podocyte processes posses MT with mixed polarity, plus-end-distal and minus-end-distal. Minus-end distal MTs are assembled in the centrosomes and then transported to cytoplasmic processes such as podocyte.28,29 In these processes, short fragments of the minus-end-distal MTs are conveyed by a kinesin-related motor protein, CHO1/MKLP1. CHO1/MKLP1 is a member of the kinesin superfamily30 and is localized in a punctuated pattern on the MTs, indicating the association of CHO1/MKLP1 with the MTs in the podocyte processes.31 Recent evidence suggested that CHO1/MKLP1 has a dual function: the generation of force during mitosis30 and transport of the MTs in the dendrites.31-34 Therefore, to establish the mixed polarity of the MTs, the podocyte expresses CHO1/MKLP1, which is essential for the formation of podocyte processes. Based on these findings, we predict that podocin may interact with kinesin, and that their interaction may influence the formation of the podocyte processes.
Second, the development of nephrotic syndrome has been associated with an early and transient induction of glomerular reactive oxygen species (ROS),35 a decrease in glomerular antioxidant enzyme (AOE) activity36 and the accumulation of the products associated with oxidative damage to membranes (lipid peroxides) in the renal cortex,37 glomeruli38,39 and urine.37-39 The injection of puromycin aminonucleoside (PAN) into rats is commonly used to cause the development of foot process effacement and proteinuria.8,40 PAN-induced podocyte injury is mediated by ROS and modulated by podocyte antioxidant defenses such as an induction of catalase, superoxide dismutase, glutathione peroxidase and so forth.41 Based on these findings, it is hypothesized that glutathion peroxidase 3 (Gpx3), a type of AOE, is related to the nephrotic syndrome and is produced within podocytes. Therefore, we speculate that Gpx3 may have a connection with podocin.
Third, the kidney actively metabolizes lipophilic molecules. Partial segments of nephrons absorb long-chain fatty acids rapidly from the blood stream, synthesize prostanoids from arachidonic acid in response to humeral factors and modulate renal function.42 Diet-induced or endogenous hyperlipidemia, in animal models of glomerular injury, accelerates the progression of glomerulosclerosis.43 Cytosolic lipid-binding proteins (LBPs) with low molecular masses (10-20 kDa), are capable of binding lipophilic molecules and are thought to play an important role in cell signaling pathways involving lipophilic molecules as well as intracellular trafficking and lipid metabolism.44,45 The LBP family includes mainly the fatty acid binding protein (FABP), acyl-CoA binding protein (ACBP), sterol carrier protein 2 (Scp2), cellular retinol binding protein (CRBP), and phosphatidylinositol transfer protein (PITP). Expression of the messenger RNA (mRNA) for these proteins is variably detected in rat glomeruli (RG) or rat mesangial cells (RMC).46 These studies led us to infer that Scp2 detected by the yeast 2-hybrid system may likely be linked to podocin in the podocytes.
Finally, a podocyte damage is considered to be an important factor for the development of glomerulosclerosis. Degradation of the glomerular basement membrane (GBM) by proteinases might be a potential mechanism involved in detachment because of the impairment of the normal process between the podocytes and the GBM. Increases in proteolytic activity may induce degradation of the GBM, which plays an important role in the progression of glomerulosclerosis. Glomerular endogenous proteinases, as well as proteinases released from the glomerular infiltrating cells are known to contribute to the degradation of the GBM.47-50 Recent studies demonstrated that glomeruli contain several proteinases, most notably metalloporteinases (MMPs) and cysteine proteinase, cathepsin L.47,49 Cathepsin B, H and L are representative cysteine proteinases found in lysosomes.51,52 These cysteine proteinases are thought to mediate the generation of bioactive products from precursor proteins.51,53 The secretion of these proteinases is stimulated by growth factors, some of which are known to contribute to the development and progression of glomerulosclerosis.54-56 The activity of the proteinases secreted from the podocytes is relevant to podocyte detachment, thereby contributing to the development and progression of glomerular sclerosis since it causes extracellular degradation of the GBM proteins.57 Therefore, cathepsin H may also be involved in the degradation of the GBM and associated with podocin.
4 candidate proteins may play an important role in renal physiology through their interaction with podocin. All of them have been implicated in nephrosis and are affected by marked podocyte changes. The fact that initiation of signal transduction by the slit diaphragm is critical for podocyte integrity and function reflects the importance of podocin to the structural integrity of the slit diaphragm complex. It remains, however, to be determined how podocin and its related proteins interact with each other in vivo, and additional studies are needed to clarify the relationship between podocin and the 4 candidate proteins identified in this study. We are currently in a process to confirm their physical interactions with podocin and clarify physiological significance of their interactions in primary podocytes. We plan to perform loss of function experiments using siRNA and gain of function experiment in heterologous system.
In conclusion, this study is a worthwhile work which presented a possibility that the interaction with podocin and candidates may cause the nephrotic syndrome.




 XML Download
XML Download