

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Autosomal dominant polycystic kidney disease (ADPKD) is a common hereditary disease, caused by mutations of either PKD1 or PKD2. End stage renal failure (ESRD) is the most dreaded complication of ADPKD. Despite recent advances in understanding the molecular pathogenesis and cystogenesis of ADPKD, and the development of ESRD due to polycystic kidney has remained elusive.1-3
The interstitial fibrosis is associated uniquely with renal failure in ADPKD4 and may be the final common pathway, in many chronic kidney diseases leading to ESRD. Renal interstitial fibroblasts are heterogeneous population that is difficult to define in situ. Recent studies have showed that epithelial-mesenchymal transition (EMT) can occur in tubular epithelial cells, which are able to migrate to the interstitium and produce extracelluar matrix protein.5-7 A recent study showed that overlap of fibroblast-specific protein 1 (FSP1)+ and α-smooth muscle actin (α-SMA)+ cells is associated with progressive fibrogenesis in animal PKD model. Some FSP1+ fibroblasts are likely derived from tubular epithelium undergoing EMT.8 In gene profiling of ADPKD, total of 87 genes were specifically regulated; 26 of these 87 genes were typical for smooth muscle, suggesting EMT as a pathogenetic factor in ADPKD.9 However, there are no direct data of EMT in human ADPKD.
Transforming growth factor-β (TGF-β) is needed to balance interactions between cells and extracelluar matrix. Studies of TGF-β signaling in the kidney had focused on the molecular biology of fibrogenesis. In recent years, TGF-β mediated glomerular and tubular epithelial cell apoptosis and EMT have been proposed as putative primary pathomechanisms that may underlie progressive renal disease.10
There are only a few data available to support the hypothesis that EMT has a role in human ADPKD. Complex mechanisms induce the EMT. However, it is not certain which factors are associated with EMT in ADPKD. To evaluate this hypothesis in human ADPKD, therefore, we immunohistochemically studied the expression of several makers of EMT.
PATIENTS AND METHODS
Patients
Informed consent was obtained from all patients. Elective transplant removal of ADPKD kidney was performed on renal graft recipients at the Samsung Medical Center. Normal renal tissue was obtained from tumor nephrectomies; tumor infiltration was excluded by histology. Tissue samples were fixed in 4% paraformaldehyde for immunohistochemistry immediately after organ removal. ADPKD kidneys from 5 patients (male : female = 3 : 2, age 54 ± 7 years) who developed ESRD and control renal tissues from 4 patients (male : female = 2 : 2, age 58 ± 9 years) with renal cell carcinoma and normal renal function were analyzed by immnunohistochemistry.
Methods
A part of each kidney was fixed in 4% paraformaldehyde and was embedded in paraffin. Sections were cut 4 µm in thickness from paraffin blocks and processed for hematoxylin-eosin and indirect immunoperoxidase staining. After deparaffinization and rehydration, the sections were treated with proteinase K and boiled with citrate buffer in microwave for antigen retrieval. After washing, they were immersed in 3% H2O2 in methanol to inhibit endogenous peroxidase and flooded with 5% bovine albumin in PBS to inhibit nonspecific reactions. Anti-α-SMA (Dako, Glostrup, Denmark), anti-E-cadherin (Zymed Lab, San Fransisco, CA, USA), anti-vimentin (Dako, Glostrup, Denmark), anti-TGF-β1 (Santa Cruz Biotech, Santa Cruz, CA, USA), and anti-Smad 2/3 (Santa Cruz Biotech, Santa Cruz, CA, USA) were applied as primary antibodies. Following primary immunoreaction, labelling was performed using streptavidin-biotin immunoperoxidase method (Nichirei Bioscience, Tokyo, Japan), and a Vectastain ABC kit (Vector Laboratories, Burlingame, CA, USA). Tissues were visualized using diaminobenzidine (DAB) as a chromogene (brown).
Images were captured by Baumer digital camera with olympus BX51 microscopy. Image analysis was accomplished using the public domain software ImageJ (available at http://rsb.info.nih.gov/ij). Operations during analysis consisted of converting the images to 8-bit grey scale [256 scales of grey, ranging from 0 (black) to 255 (white)], plotting the distribution of grey values, thresholding and measuring total area of positive cells automatically. For manual counting of positive tubules, several randomly selected fields, totaling 0.25 mm2 from the center area of the well were used. Positive tubules (%) were the percent of positive tubules/total tubules (× 200).
All results were analyzed using SPSS 12.0 and expressed as mean ± standard deviation. Statistical significances (p < 0.05) were analyzed by using the Mann-Whitney test.
RESULTS
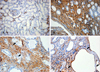
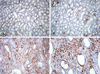
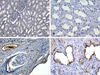
There were severe interstitial fibrosis and proliferation of α-SMA+ myofibroblasts in ADPKD compared to control kidneys. In ADPKD, tubular epithelial cells of dilated cysts were occasionally α-SMA+ (Fig. 1). E-cadherin is an epithelial cell marker. There were some portions with decreased expression in cystic epithelial cells of large renal cysts in ADPKD (Fig. 2). Vimentin is a mesenchymal cell marker, and it's vimentin expression was increased in the stromal and cystic epithelial cells in ADPKD kidneys (Fig. 3). TGF-β1 and Smad 2/3 were regulated upward in cystic epithelial cells (Figs. 4 and 5).
These immunohistochemical findings were quantitatively summarized by computer-assisted image analyzer and positive tubules (%) (Table 1). There were significant increases of α-SMA (34.3 ± 11.7 vs 0.9 ± 1.5), vimentin (19.9 ± 3.9 vs 3.3 ± 1.4), TGF-β1 (5.42 ± 2.83 vs 0) and Smad 2/3 (3.4 ± 1.7 vs 0.7 ± 0.6) in ADPKD kidneys than control kidneys, demonstrated by computer-assisted image analyzer. When we analyzed the positive tubules (%), the results were the same as computer-assisted image analyzer. Large cystic tubular cells in ADPKD showed many α-SMA+, vimentin++ and E-cadherin- (Table 2).
DISCUSSION
Human ADPKD is a heterogenic genetic disorder that progresses to ESRD. Progression to ESRD in ADPKD is associated with not only forming cysts, but also the accumulation of interstitial collagens, presumably because of proliferation of fibroblasts.4,11 As cysts arise from only a small percentage of nephrons, other downstream mechanisms are important in progression to ESRD in ADPKD. In our study, proliferation of α-SMA+ myofibroblasts and interstitial fibrosis were key features of ADPKD with ESRD. α-SMA is normally synthesized by vascular smooth muscle cells and myofibroblasts differ from fibroblasts in granulation tissue. The origin of contractile myofibroblasts has not been easy to characterize. The conventional view is that these myofibroblsts are originated by differentiation of resident renal fibroblast, or migration of pericytes, or tubular epithelia cells by EMT.
EMT and transdifferentiation are concepts which originally belong to embryology and oncology. Cell shifting between epithelial and mesenchymal phenotype, by turning on and off specific genes during development, is a well recognized mechanism that characterizes the embryonal plasticity.12 EMT can also play a key element in metastasis of tumors derived from epithelial tissues.13 Recently emerging evidences suggest that EMT can be an important role in several glomelulonephritis,14,15 peritoneal fibrosis of peritoneal dialysis,16 failing renal transplants17 and diabetic nephropathy.2 However, there are few data of EMT available in human ADPKD.
In a recent study by Okada et al.,8 many nonoverlapping as well as fewer overlapping populations of FSP1+ and α-SMA+ cells shared in the collagen expression were found to be associated with progressive fibrogenesis in pcy mice undergoing cystogenesis. Some FSP1+ fibroblasts are likely derived from tubular epithelium undergoing EMT, whereas α-SMA +, VIM-cells probably represent vascular smooth muscle cells or pericytes surviving vessel attenuation during the chaos of fibrogenesis. A recent study by Schieren et al.9 showed that total of 87 genes in gene profiling of ADPKD were specifically regulated; 26 of these 87 genes were typical for smooth muscle, suggesting EMT as a pathogenetic factor in ADPKD. They showed that smooth muscle actin and caldesmon were mainly expressed in the interstitium of ADPKD kidneys. In our study, tubular epithelial cells in ADPKD lost epithelial marker (E-cadherin) and expressed mesenchymal markers (α-SMA, vimentin). These finding suggest that EMT has an important role in progression in ADPKD.
Since the discovery of TGF-β as a key mediator of glomerular and tubulointerstitial fibrosis in many chronic kidney diseases over a decade ago, studies of TGF-β signaling had been focused on the cell proliferation and extracellular-matirx synthesis.18 In recent years, TGF-β signaling was found to play central role in renal fibrosis19 by EMT.20,21 In the present study, we showed that TGF-β-Smad signaling was regulated upward in ADPKD, thus suggesting that TGF-β-Smad signaling is associated with renal EMT and renal fibrosis in ADPKD. It is highly likely that cyst cell proliferation and fluid secretion initiated the renal cysts, that enlarged cyst then stretched tubular cells that would secrete the TGF-β, and that increased TGF-β mediated EMT and interstitial fibrosis. In future studies, therefore, we plan to suppress TGF-β and EMT to delay the clinical course to ESRD in ADPKD.
In summary, α-SMA+ myofibroblasts and interstitial fibrosis were key features of ADPKD with ESRD. Cystic tubular epithelial cells lost epithelial marker and expressed mesenchymal markers. TGF-β-Smad signaling was regulated upward in ADPKD, suggesting that TGF-β mediated EMT has a role in progression of ADPKD.




 XML Download
XML Download