

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Despite uninterrupted improvements in fetal monitoring and neonatal intensive care medicine, perinatal hypoxic-ischemic encephalopathy still remains an important cause of neonatal mortality and permanent neurological sequelae such as cerebral palsy, mental retardation, learning disabilities, and epilepsy.1 At present, there is no clinically efficacious treatment available for this common disorder. Therefore, the development of a new therapeutic modality to improve the prognosis of this disease is an urgent issue.
Recently, the neuroprotective effects of exogenously administered G-CSF have been reported in several adult animal models of ischemic brain injury2-8 as well as in phases I/II clinical studies of stroke.9,10 The neuroprotective effects exerted by G-CSF are known to be mediated by a multitude of mechanisms,11 including inhibition of apoptosis,6 anti-inflammatory effects,3 and stimulation of endogenous stem cell proliferation.7,8 G-CSF is one of the few growth factors that have been approved for clinical use to treat neutropenia, even in newborn infants.12,13 Therefore, any favorable experimental results can readily be translated into clinical practice. However, results obtained in adults cannot necessarily be extrapolated into neonatal medicine due to the dramatic differences in maturational stage and pathophysiology of perinatal and adult brains. Up to now, only 2 studies have reported the effects of G-CSF in neonatal brain injury. While its beneficial effects have been shown in one study,14 G-CSF was found to be detrimental to neonatal brain injury in the other.15 Therefore, the role of G-CSF in neonatal brain injury is still controversial, and further studies are necessary to clarify this.
This study was undertaken to determine whether G-CSF could reduce cerebral injury following HI in the developing brain. As we have already shown in our previous studies,16,17 apoptosis plays a major role in the development of hypoxic-ischemic neonatal brain injury. In the present study, we tested whether G-CSF is neuroprotective by inhibiting apoptosis, thereby reducing the ensuing cerebral infarction in a newborn rat pup model of cerebral HI. Apoptosis was identified by TUNEL staining and flow cytometry with a combination of fluorescinated annexin V and propidium iodide (PI) and JC-1 (5,5',6,6'-tetrachloro-1,1',3,3'-tetraethylbenzimidazolyl-carb ocyanine iodide). The extent of cerebral infarction was evaluated at 2 weeks after HI to determine whether the neuroprotective effect, if any, of G-CSF was transient or sustained.
MATERIALS AND METHODS
Hypoxia-ischemia
The experimental protocols described herein were reviewed and approved by the Animal Care and Use Committee of Samsung Biomedical Research Center, Seoul, Korea. This study was performed in accordance with the institutional and National Institutes of Health guidelines for laboratory animal care. Seven-day-old Sprague-Dawley male rats (Daihan Biolink Co., Seoul, Korea) weighing 13 - 16 g were used for the experiments to exclude any gender-related differences in brain injury.18 The P7 male rat pups (n = 56) were randomly divided into 3 groups: (1) normoxia control group (NC, n = 18); (2) HI control group (HC, n = 16); and (3) HI with G-CSF treatment group (HG, n = 22). Cerebral HI was induced by a modification of the method16,17 originally reported by Rice et al.19 The neck was incised in the midline, and the right common carotid artery was permanently ligated with 4 - 0 silk under methoxyflurane anesthesia. After 2 hours of recovery period, the animals were exposed to 110 minutes of hypoxia (8% O2/92% N2) by placing them in airtight containers that had been partially submerged in a 37℃ water bath to maintain a constant thermal environment. Pups in NC received a Sham operation without carotid artery ligation. Animals in HG received an intraperitoneal injection of G-CSF (a kind gift from Donga Pharm. Co., Seoul, Korea) at a dose of 50 µg/kg of body weight immediately after the hypoxic insult,8 or an equal volume of normal saline as a NC and HC. Then, the pups were returned to their dams and sacrificed at 24 hours after HI for TUNEL stain and flow cytometry (n = 11 in NC, 7 in HC, and 11 in HG) under deep pentobarbital anesthesia (60 mg/kg, intraperitoneal). The remaining rat pups (n = 7 in NC, 9 in HC, and 11 in HG) were weighed daily for 2 weeks after HI until sacrifice for brain volume measurements.
Preparation of brain specimens
Fresh whole brain tissue obtained at 24 hours after HI was placed on a glass petri dish on ice containing 1 - 2 mL of phosphate-buffered saline (PBS), and transected in the coronal plane at the level of the mid-dorsal hippocampus.20 The anterior portion of the right cerebral hemispheres was processed for TUNEL staining, and the posterior portion of the cerebral hemisphere within the middle cerebral artery territory, approximately 100 mg/block, was dissociated into a single cell and used for flow cytometric measurements.16,17
TUNEL staining
The extent of DNA fragmentation was delineated by analyzing brain tissue obtained from the anterior half of the ipsilateral cerebral cortex using a peroxidase in situ apoptosis detection kit S7100 (Chemicon International, Temecula, CA, USA) at 24 hours after HI. TUNEL-positive cells were identified by an optical microscope with 400 high power fields. Ten images were taken from each sample and the number of TUNEL-positive cells on a high power field was determined.
Flow cytometry
To evaluate the extent of apoptotic and necrotic cells, the posterior portion of the ipsilateral cerebral cortex was dissociated into a single cell and flow cytometry was done with a combination of PI (Sigma, St. Louis, MO, USA) and annexin V-FITC (fluorescein isothiocyanate) (Pharmingen, San Diego, CA, USA). Flow cytometric analysis was performed by PAS (Particle Analyzing System, Partec, Münster, Germany) equipped with an argon ion laser tuned at 488 nm wavelength. Green FITC-annexin V fluorescence was measured at 530 ± 15 nm, and red PI fluorescence was measured at 600 nm.16,17
Mitochondrial membrane potential was estimated using fluorescent probe JC-1 (Molecular Probes, Eugene, OR, USA).21 The dissociated cortical cell suspensions were adjusted to the density of 1 × 106 cells/mL and stained for 20 minutes with 2.0 µg/mL of JC-1 at 37℃. JC-1 was excited with the 488 nm argon laser, and JC-1 green and orange fluorescences were recorded on FL1 (530 ± 15 nm band pass filter) and FL2 (575 ± 13 nm band pass filter) channels.
Brain volume measurement
After transcardiac perfusion with 0.1 M of PBS, the brains were carefully removed at 2 weeks after HI and blocked with paraffin after overnight fixation with 4% paraformaldehyde. The 5 µm thick serial sections were made at an interval of 100 µm and stained with hematoxylin-eosin for volumetric analysis.
All volumetric qualifications were performed with an Olympus BX 40 photomicroscope (Olympus Optical Co., Ltd., Tokyo, Japan) equipped with a high resolution CCD camera, a motorized XYZ axis computer-controlled stage, and Stereoinvestigator software package (ver. 6.52, Micro Bright Field, VT, USA). When calculating volume, the cross-sectional areas of the region of interest (ROI) in each section were traced on the computer screen at low power with a 1.5 ×/4 × lens, and volume of the ROI was calculated by using the Stereoinvestigator software according to Cavalieri's principle.22
Using this sampling strategy, approximately 8 histology sections per brain in the affected and control hemisphere in each animal were evaluated for hemispheric measurements. For anatomical evaluation of the extent of cerebral injury, the ratio of the ipsilateral remaining cerebral hemisphere volume to the volume of the corresponding contralateral cerebral hemisphere is expressed as a percentage. An examiner blind to the treatment group quantified.
Statistical analysis
All data are expressed as mean ± standard deviation. Kruskal-Wallis test was used for multiple group comparisons followed by Wilcoxon rank sum test including Bonferroni adjustment for a comparison between 2 groups. All statistical analyses described above were carried out using the SAS Enterprise Guide version 3 (SAS Institute, Cary, NC, USA). A p value of < 0.05 was considered significant.
RESULTS
TUNEL staining
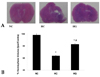
In the ipsilateral anterior portion of the cerebral cortex at 24 hours after HI significantly increased numbers of TUNEL-positive cells were observed in HC compared to NC (59.6 ± 9.2 vs. 0.2 ± 0.1, p < 0.01). However, this increase in the number of apoptotic cells was significantly attenuated in HG compared to HC (14.3 ± 16.9 vs. 59.6 ± 9.2, p < 0.05) (Fig. 1).
Flow cytometry
Representative analyses and regional percentage of an annexin V versus PI dot plot of the ipsilateral cerebral cortex in NC, HC, and HG at 24 hours after HI are presented in Fig. 2A. In HG, the increased percentage of apoptotic cells in Q4 (annexin V+/PI-) and decreased percentage of live cells in Q3 (annexin V-/PI-) observed in HC were significantly improved but the attenuation of secondary necrotic cells in Q2 (annexin V+/PI+) did not reach statistical significance, and the damaged cells in Q1 (annexin V-/PI+) were not significantly different from those in HC (Table 1).
Body weight gain
Significantly retarded body weight gain was observed in HC compared to NC, starting from 3 days after HI (p < 0.05). The weight gain, which is indicative of general well being, was significantly improved in HG from 10 days after HI compared to HC (p < 0.05) (Fig. 3).
Measurement of brain volume
In HC, there was a significant decrease in per centage of intact ipsilateral hemispheric volume compared to NC (p < 0.01) at 2 weeks after HI injury. The HG group showed a marked protective effect and significantly increased percentage of ipsilateral hemispheric volume compared to the HC group (p < 0.01) 2 weeks after HI (Fig. 4).
DISCUSSION
In the present study, G-CSF significantly attenuated the HI-induced loss of body weight and increased apoptosis and ensuing cerebral infarction in the newborn rat. In accordance with our data, Yata et al.14 have also reported the neuroprotective effects of G-CSF in neonatal-hypoxic ischemic brain injury. The safety and feasibility of G-CSF have been demonstrated in recent human stroke therapy trials.9,10 Furthermore, G-CSF is already available clinically in neonates.12,13 Taken together, these findings strongly support the potential use of G-CSF as therapeutic agents in the clinical setting of perinatal hypoxic ischemic encephalopathy where effective treatments have not yet been established.
Although the mechanisms by which G-CSF serves a neuroprotective agent have not fully been clarified, G-CSF may act in a coordinated fashion at multiple levels,11 including inhibition of apoptosis,6 modulation of inflammation,3 and recruitment of endogenous stem cells.7,8 In the present study, TUNEL staining was performed to detect nuclear DNA breakdown, a common feature of apoptosis.23 Flow cytometry using annexin V was done to detect exposure of phosphatidylserine outside of the plasma membrane, the early and most characteristic change of apoptosis.24,25 PI was used to detect the disruption of the plasma membrane and alterations in permeability observed in necrotic cells. Decreased mitochondrial membrane potential, observed during early apoptosis, were measured by counting the shift of JC-1 fluorescence from orange to green with flow cytometry.21 Together, our data on HI-induced increase in TUNEL-positive cells, apoptotic (annexin V+/PI-) cells, and JC-1 fluorescence shifted from orange to green support the assumption that apoptosis is the primary mode of cell death following HI in the developing brain.16,17,26 Our data of significant attenuation of these abnormalities with G-CSF treatment also support the assumption that the neuroprotective action of G-CSF is mediated mainly by inhibition of apoptosis in a newborn rat model of cerebral HI.14
In the present study, a single dose of G-CSF given immediately after HI significantly attenuated the cerebral infarction at 2 weeks after HI. These results implicate that the neuroprotective effects of G-CSF are not transient. In our previous studies,16,17 we have shown that apoptosis plays an important role in the development of delayed infarction, and that the inhibition of apoptosis with cycloheximide significantly attenuates ensuing cerebral infarction in the newborn rat model of cerebral HI. Therefore, our data on significant reduction of infarction with G-CSF treatment strongly suggest that G-CSF given immediately after HI could attenuate and prevent the activation of subsequent neurotoxic, probably apoptotic cascade, ultimately leading to irreversible neuronal cell death and delayed cerebral infarction occurring days or months later.26-29
In the present study, G-CSF was given immediately after HI. However, as most asphyxial events occur before birth and enrollment of newborns for clinical trials can take at least several hours for a mother to recover from a delivery and to give informed consent, it is practically impossible to start any therapy immediately after birth. There is a danger that delay in applying potentially brain-saving treatments beyond the therapeutic window might abolish or drastically reduce its therapeutic effectiveness.17,27-29 Therefore, the long therapeutic window of G-CSF reported in both preclinical8 and clinical studies of adult stroke9,10 suggests therapeutic potential of this agent in humans with perinatal hypoxic-ischemic encephalopathy. Further studies on dosage, therapeutic window, and safety in neonates are necessary for the successful translation of the benefits of G-CSF treatment observed in this animal study into clinical trials.
In summary, G-CSF was demonstrated to have neuroprotective effects by inhibiting apoptosis, reducing the ensuing cerebral infarction in a newborn rat pup model of cerebral HI. Our data suggest that G-CSF is a potentially novel therapeutic agent in perinatal hypoxic-ischemic encephalopathy, and further clinical studies are warranted.




 XML Download
XML Download