

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Multiple endocrine neoplasia type 1 (MEN1) is an autosomal dominant hereditary disorder. The clinical diagnosis of MEN1 can be made in a patient who has tumors in at least two of the following tissues: pituitary, parathyroid, and pancreatic islet cells. In addition, MEN1 carriers can have adrenal or thyroid adenomas, bronchial or thymic carcinoid tumors, and non-endocrine tumors, such as lipomas, angiofibromas, collagenomas, and leiomyomas.1-4 In Korean literature, Oh et al. reported the first case in 1986, diagnosed radiologically, and a total of 24 cases have been reported since. Nevertheless, MEN1 cases with accompanying multiple leiomyoma have not yet been reported in Korean literature.5-18 Recently, a case of MEN1 was identified with accompanying leiomyomas in the bladder, the uterus, and the esophagus, in addition to typical endocrine tumors, such as parathyroid adenoma, pituitary adenoma, pancreatic tumor, and adrenal cortical tumor. This report describes this MEN1 patient with multiple leiomyomas, who revealed a novel germline missense mutation, D350V, in the MEN1 gene, with a review of the relevant literature.
CASE REPORT
A 50-year-old female presented with chronic fatigue. The patient was diagnosed with osteoporosis five years prior and was taking alendronate. Hypercalcemia was diagnosed two years following the osteoporosis diagnosis, and the patient visited the hospital for a comprehensive check-up. The patient did not have any history of diabetes, hypertension, tuberculosis, or hepatitis. She was the first daughter of two sons and two daughters. Concurrently, her 81-year-old mother, who was bedridden for unknown reasons, was also undergoing tests for hypercalcemia, and was subsequently diagnosed with multiple parathyroid adenomas and pancreatic tumors, based on the test results. Her father had a history of sudden death by an acute cardiac event.
In the neck examination, the thyroid was not enlarged, and palpable tumors were absent. Heart and lung sounds were normal, and no abnormalities were detected in the abdomen, all extremities, or neurological tests. The axillary hair and the pubic hair were normal, hirsutism was absent, and galactorrhea was not observed.
In blood electrolyte tests, sodium was 144 mmol/L, potassium was 4.0 mmol/L, chloride was 107 mmol/L, blood calcium was 12.6 mg/mL, ionized calcium was 5.4 mg/dL, and inorganic phosphate was 3.3 mg/dL. In general biochemical tests, fasting blood glucose was 93 mg/dL, blood urea nitrogen was 8.9 mg/dL, creatinine was 0.8 mg/dL, and the hepatic function test was normal. Her 24-hour urinary calcium excretion was 326 mg/day, the amount of phosphate excretion was 460 mg/day, and the creatinine clearance rate was 64 mL/min. Thyroid hormone tests were within normal range, and intact-PTH was increased to 99.8 pg/mL. Prolactin was also mildly increased to 27.63 ng/mL. Plasma insulin was 9.2 uIU/mL (4.2 - 48), gastrin was 21 pg/mL (0 - 90), glucagon was 40 pg/mL (40 - 130), pancreatic polypeptide was 79 pmol/L (< 100), and vasoactive intestinal polypeptide was 32 pg/mL (< 100). In dual X-ray absorptiometry (DXA), the T score of the femur was decreased to - 2.0, confirming the osteopenia finding.
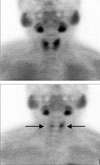
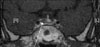
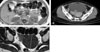
In the neck ultrasound, in the posterior area of the left thyroid, a tumor suspected to be a parathyroid adenoma 10 mm in size was detected, and in the parathyroid scan, uptake was detected in two glands (Fig. 1). In the neck computed tomography (CT), parathyroid tumors, 10 mm in the left side and 8 mm in the right side, were detected. In the chest CT, abnormal findings in the lung and the mediastinum were not detected. On the brain magnetic resonance imaging (MRI), a tumor 7 mm in size in the pituitary was detected (Fig. 2). On the abdominal MRI, a 2.3 cm left adrenal tumor and a pancreatic tumor were observed, and a 7 cm uterine myoma and a bladder wall myoma were detected (Fig. 3). Esophagogastroduodenoscopy (EGD) and endoscopic ultrasonography (EUS) revealed an esophageal leiomyoma 20 mm in size in the area 20 cm away from the incisor, and in the pancreatic head, a tumor 6 mm in size was detected (Fig. 4). In general positron emission tomography (PET), increased fluorodeoxyglucose (FDG) uptake in the pituitary was seen. Nonetheless, the increase of the uptake by other lesions was not detected, and thus indicated the benign nature of the multiple tumors.
From the peripheral blood of the patient and her mother, genomic DNA was extracted, and sequencing of the MEN1 gene was performed. Sequencing analysis revealed a transition of GAG to GTG at codon 350 of exon 7, causing an amino acid change from aspartic acid to valine, D350V (Fig. 5).
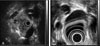
Because of the detection of double parathyroid adenoma, pituitary adenoma, pancreatic tumor, and adrenal cortical tumor, as well as accompanying multiple leiomyomas in the bladder, the uterus, and the esophagus, MEN1 was diagnosed. For parathyroid adenoma, a subtotal parathyroidectomy was performed, leaving half of the left inferior parathyroid gland, and a prophylactic thymectomy was performed as well. In postsurgical histological tests, the diagnosis of parathyroid adenoma was confirmed (Fig. 6). After surgery, the intact-PTH level was decreased to 5.79 pg/mL. The remaining tumors are under follow up observation.
DISCUSSION
MEN1 can be diagnosed by the identification of two or more of the following: parathyroid tumor, pancreatic islet cell tumor, and pituitary adenoma. In addition, MEN1 may also include a carcinoid tumor, lipoma, adrenal gland tumor, adrenal hyperplasia, and leiomyoma.1-4 It is inherited autosomal dominantly in most cases, and occurs in familial or sporadic forms. In Korea, approximately 24 cases have been previously reported, and among them, 11 cases were familial, and cases with accompanying leiomyoma have not been reported.5-18 The clinicopathological features of MEN1-associated tumors are generally similar to those of tumors arising sporadically in the same tissue; nevertheless, MEN1-associated tumors show a distinct pattern in three aspects.4 Firstly, MEN1 develops in several endocrine tissues and is multicentric. Due to its multicentric characteristic, its recurrence rate after surgery is high. Secondly, some MEN1 tumors (parathyroid and gastrin cell) typically present approximately one or more decades earlier than sporadic tumors of the same tissue. Thirdly, some MEN1 tumors have the potential for malignancy, and the pertinent malignant tumors have been known to be a major cause of death in MEN1 patients. The parathyroid is the most prevalent endocrine organ affected, and parathyroid involvement is seen in 90% of patients.1 In Korea, a tumor in the parathyroid was seen in 83% of cases. In nine of these cases, pathological findings were reported in the literature, and 56% (5 cases) were parathyroid hyperplasia, and 44% (4 cases) were parathyroid adenoma (Table 1).5-7,9-10,12-14,16-18
Pancreatic islet cell tumors are detected in approximately 30 - 80% of MEN1 cases, and these could be either functional or non-functional. In the cases of functional tumors, it has been reported that various hormones could be released. In Korean MEN1 cases, a pancreatic islet cell tumor was developed in 96% of the cases. Cases secreting insulin (32%, 8 cases) and gastrin (20%, 5 cases) were most prevalent among functional tumors. The cases considered to be non-functional were 16% (4 cases) (Table 1).5-18
Pituitary tumors, most commonly prolactinomas, are present in 10 - 50% of symptomatic MEN1 patients. In Korean cases, a pituitary tumor was detected in 75% of the cases, and among them, prolactin-secreting adenoma was the most prevalent, making up 68% (13 cases) (Table 1).5,7-9,11,13-15,17,18
Other so-called non-classical neoplasms occur frequently in MEN1 patients. Adrenal cortical tumors are detected in approximately 40% of Korean patients. The majority of these tumors were bilateral, hyperplastic and non-functional, and caused minimal morbidity. In addition to endocrine tumors, non-endocrine mesenchymal tumors, such as lipoma, angiofibroma, collagenoma, and leiomyoma, have been reported to be associated with MEN1 and mutations in the MEN1 gene.2,19-21 Leiomyoma has been reported to develop in the esophagus, lung, uterus, skin, and ureter, and the difference from sporadic leiomyoma is its multicentric development in many cases.2,3 In Korean reports, non-endocrine tumors have only been reported in three cases: carcinoid tumors (two cases) and leiomyomas (the current study).5-8,14
The MEN1 gene is localized to chromosome 11q13, was identified by positional cloning, and is composed of 10 exons.22,23 It encodes a 610 amino acid protein, menin, which is localized primarily to the nucleus.23 Menin has a tumor-suppressor function that involves direct binding to the JunD protein and the inhibition of JunD-dependent transcription.24 Mutation sites in the MEN1 gene are found throughout the entire coding region. A genotype-phenotype correlation has not been clearly established for the mutations, although it has been suggested that some of the mutations might be associated with the formation of specific tumors.25
In Korea, genetic analysis has been reported in only 38% of MEN1 patients. This analysis is summarized in Table 1, and a frameshift mutation was detected in two cases, a nonsense mutation in two cases, and a missense mutation in three cases. In our case, a germline missense mutation, D350V, was identified. To our knowledge, this mutation has not been reported to date.18,26-30
The first DNA analysis of a leiomyoma of the esophagus in a patient with MEN1 failed to detect allelic deletions at the MEN1 locus.19 This implied that the tumor arose through a completely independent mechanism not associated with the patient's underlying MEN1 disease. However, in a study conducted subsequently, loss of heterozygosity (LOH) at the MEN1 locus was shown in two esophageal leiomyomas from a MEN1 patient.20 Meanwhile, another study reported that the MEN1 gene contributes to the development of multiple esophageal and uterine leiomyomas in MEN1 patients and that the gene does not play a significant role in the tumorigenesis of sporadic uterine leiomyoma.2 Thus, leiomyomas in MEN1 and sporadic patients most likely develop through different mechanisms.
In conclusion, although leiomyomas in MEN1 have not previously been reported in Korea, they should be considered an integral part of MEN1 and examined carefully. Additionally, we found a novel mutation, D350V, in exon 7 of the MEN1 gene.



 XML Download
XML Download