

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
It is well known that TZDs (rosiglitazone, troglitazone, and pioglitazone) are insulin-sensitizing agents that possess anti-diabetic activities in vivo. TZDs are ligands of peroxisome-proliferator-activated receptor (PPAR), a member of a larger family of ligand-activated nuclear receptor transcription factors. Recent evidence suggests that TZDs exert potent anti-proliferative and anti-inflammatory effects on the vascular wall by inhibiting VSMC proliferation, monocyte inflammatory cytokines, macrophage activation, and the expression of cell adhesion molecules.1-6 Furthermore, TZDs are known to inhibit VSMC proliferation by diminishing mitogen-induced degradation of p27Kip1, thereby resulting in increased activity of RB.7 However, the upstream signaling mechanisms associated with the abovementioned mechanism have not yet been elucidated. The mammalian target of rapamycin (mTOR) is an enzyme located in the non-particulate region of the cytoplasm that plays an essential role in connecting extracellular signals with intracellular pathways that regulate cell cycle and proliferation. Rapamycin eluting stents have proven to significantly reduce the rate of coronary stent restenosis in human clinical trials.8 Through inactivation of p70S6 kinase (P70S6K) and eukaryocytic initiation factor 4E (eIF4E), rapamycin increases the activity of p27Kip1 and RB.9,10 The mTOR itself is known to be activated by the phosphoinositide 3-kinase (PI3K)-Akt pathway.11,12
We used insulin as a mitogenic stimulus because of the important role of hyperinsulinemia in the development of neointimal hyperplasia, the concomitant inducer of translation initiation factor activity (phosphorylation of 4EBP-1 and P70S6K), and critical cell cycle regulators downstream of mTOR. Insulin and angiotensin II are known to activate the PI3K/Akt/mTOR/P70S6K pathways and to phosphorylate 4E-BP1.13-15
In this study, we hypothesized that rosiglitazone, a peroxisome proliferator-activated receptor-γ (PPAR-γ) agonist, inhibits insulin-stimulated VSMC proliferation through the inhibition of the Akt-mTOR-P70S6K system, and that the Akt-mTOR-P70S kinase system is a specific major pathway for the inhibition of VSMC proliferation.
MATERIALS AND METHODS
Rosiglitazone was a generous gift from Glaxo SmithKline. All other materials were purchased from commercial suppliers. Spontaneous diabetic OLETF rats were kindly supplied by the Tokushima Research Institute, Otsuka Pharmaceutical, Tokushima, Japan. Four-week-old male OLETF male rats (70 - 80 g initial weight) were housed in a temperature-controlled environment under a 12-hour light/dark cycle and were given standard chow and water ad libitum. Animals were handled in compliance with the Guiding Principles in the Care and Use of Animals. All rats were fed standard rat chow until they were 28 weeks of age. Rosiglitazone (GlaxoSmith Kline; 3 mg/kg), was mixed with pulverized standard chow to a final concentration of 0.015% (wt/wt), and rats were treated for 1 week. After 3 weeks, carotid artery balloon injury was performed.
Primary culture of RAoSMCs
RAoSMCs were isolated and purified by the methods previously described.16 The thoracic aortas from 6- to 8-week-old Sprague-Dawley rats were removed and transferred to serum-free Dulbecco's modified Eagle medium (DMEM; Invitrogen Co., Carlsbad, CA, USA) containing 100 U/mL of penicillin and 100 g/mL of streptomycin. The aorta was freed from connective tissue, transferred into a petri dish containing 5 mL of an enzyme dissociation mixture containing DMEM with 1 mg/mL of collagenase type I (Sigma, St. Louis, MO, USA) and 0.5 µg/mL of elastase (USB Bioscience, Cleveland, OH, USA), and incubated for 30 minutes at 37℃. The aorta was then transferred into DMEM and the adventitia was stripped off with forceps under a binocular microscope. The aorta was transferred into a plastic tube containing 5 mL of the enzyme dissociation mixture and incubated for 2 hours at 37℃. The suspension was centrifuged (1500 rpm for 10 minutes), and the pellet was resuspended in DMEM with 10% fetal bovine serum (FBS). RAoSMCs were cultured in DMEM supplemented with 10% FBS, 100 IU/mL of penicillin, and 100 g/mL of streptomycin in 75-cm2 flasks at 37℃ in a humidified atmosphere of 95% air and 5% CO2 (Forma Scientific, Inc., Marjetta, OH, USA).
Proliferation assay
RAoSMCs were plated in triplicate in wells of 96-well plates at a density of 1 × 104 per well and treated with rosiglitazone prior to insulin stimulation. Cell viability was determined by MTT assay. After the incubation period, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution (Sigma) was added to each well to a final concentration of 0.5 mg/mL and the mixture was incubated at 37℃ for 3 hours to allow MTT reduction. The formazan crystals were dissolved by adding dimethylsulfoxide (DMSO) and absorbance was measured at 570 nm with a spectrophotometer.
Immunoblot analysis
RAoSMCs were washed once in PBS and lysed in a lysis buffer (Cell Signaling, Beverly, MA, USA) containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM Na2-EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 mg/mL leupeptin, and 1 mM PMSF. Protein concentrations were determined using the Bradford protein assay kit (BioRad, Hercules, CA, USA). Proteins were resolved on 12% SDS-polyacrylamide gel and transferred to PVDF membrane (Millipore Co., Bedford, MA, USA). After blocking the membrane with Tris-buffered saline-tween 20 (TBS-T, 0.1% tween 20) containing 5% nonfat dried milk for 1 hour at room temperature, the membrane was washed twice with TBS-T and incubated with primary antibody for 1 hour at room temperature or overnight at 4℃. The membrane was washed 3 times with TBS-T for 10 minutes each and then incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 hour at room temperature. After extensive washing, the bands were detected by enhanced chemiluminescence (ECL) reagent (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The band intensities were quantified using Photo-Image System (Molecular Dynamics, Uppsala, Sweden). For treatment with PPAR-g ligand, sub-confluent RaoSMCs (passage 4 and 8) were made quiescent by serum starvation (0.1% FBS) for 3 days. The cells were treated with 10 µM/L of rosiglitazone for 24 hours before the addition of insulin (1 µM). Fresh preparations of VSMCs were used for each experiment.
Perfusion of vessels and H&E staining
OLETF rats treated with balloon injury were sacrificed 3 weeks after the injury, and their carotid arteries were excised. The heart was perfused with Krebs-Ringer Bicarbonate Buffer (KRBB : 120 mM NaCl, 25 mM NaHCO3, 5 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4, 2.5 mM CaCl2, and 20 mM MOPS) for 10 minutes to wash out the blood and then fixed with 10% formalin. The carotid artery was sliced transversely. Within 24 hours of fixation, each section was embedded in paraffin. Serial 2 µm-thick carotid artery sections were cut with a microtome and mounted on siliconized slides. Paraffin sections were stained with hematoxylin and eosin (H&E) and evaluated the histological effects with light microscopy. Normal and neointimal areas were measured with NIH Image.
Histology and immunohistochemistry
OLETF rats treated with balloon injury were sacrificed and their carotid arteries were excised. The vessels were perfusion-fixed with 10% (v/v) neutral buffered formaldehyde for 24 hours, transversely sectioned into serial thick sections, and embedded in paraffin by routine methods. Sections of 2 µm in thickness were mounted on gelatin-coated glass slides to ensure that different stains could be used on successive sections of tissue cut through the areas of ballon injury. After deparaffinization and rehydration, the sections were analyzed with rabbit anti-p-mTOR (Cell Signaling Technology). Texas Red-conjugated goat anti-rabbit IgG (Jackson ImmunoResearch Lab, PA, USA) was used as secondary antibody. All images were made by using an excitation filter under reflected light fluorescence microscopy and transferred to a computer equipped with MetaMorph software v. 4.6 (Universal Imaging Corp., PA, USA).
Induction of neointima formation after balloon denudation
Balloon denudation of the common carotid artery endothelium was evoked in OLETF rats. Under ketamine (10 mg/kg, Yuhan, Seoul, Korea) and Xylazine (5 mg/kg, Bayer Korea, Seoul, Korea) anesthesia, a neck midline incision was made and after exposure of the left carotid artery, a 2F Fogarty balloon catheter (Edwards Lifesciences, Mississauga, ON, Canada) was inserted into the external carotid branch to the aortic arch, insufflated to produce slight resistance, and withdrawn 3 times. They were compared to sham-operated controls in which the same procedure was performed except for balloon insertion. Animal housing and experimentation were in accordance with Animal Care and NIH guidelines and approved by the local animal care committee.
RESULTS
Anti-proliferative effects of rosiglitazone in insulin-stimulated RAoSMC
In order to investigate whether rosiglitazone affects RAoSMC proliferation induced by insulin treatment, RAoSMCs were treated with 1 µM of insulin in the presence or absence of rosiglitazone, and a cell proliferation assay was performed. Fig. 1A shows that cell proliferation gradually increased within 3 days upon treatment with insulin, but 10 µM of rosiglitazone inhibited the augmentation of cell proliferation induced by insulin treatment, suggesting that rosiglitazone affects cell proliferation due to insulin treatment.
Inhibitory effects of rosiglitazone on activation of mTOR in insulin-stimulated RAoSMCs
To confirm the inhibitory effect of various concentrations of rosiglitazone on p-mTOR, changes in p-mTOR levels were estimated by immunoblot analysis. Thus, RAoSMCs at 70% confluence were made quiescent by serum starvation with 0.1% FBS for 3 days. Cells were then treated with various concentrations of rosiglitazone before stimulation with 1 µM of insulin. As shown in Fig. 1B, the activation of mTOR upon insulin stimulus was significantly inhibited by rosiglitazone in a dose-dependent manner. Compared to cells treated with insulin alone, cells treated with rosiglitazone before insulin stimulus demonstrated significant inhibition of phosphorylated mTOR.
Inhibitory effects of rosiglitazone on mTOR activation in balloon-injured OLETF rats
We then performed immunohistochemistry to determine the activation of mTOR during neointima expansion in OLETF rats. The inhibitory effect of rosiglitazone was detected not only in vitro but also in vivo. As shown in Fig. 2, there was no immunoreactivity for p-mTOR in normal uninjured OLETF rats. In response to vascular injury, the expression of p-mTOR was increased after balloon injury due to proliferation of RAoSMCs. However, the intensity of p-mTOR immunostaining was markedly decreased in OLETF rats when treated with rosiglitazone after balloon injury compared to balloon-injured OLETF rats with no rosiglitazone treatment.
Akt-mTOR-P70S6K signaling pathway in RAoSMCs treated with rosiglitazone
Binding of insulin to its receptor activates the receptor tyrosine kinase that phosphorylates the insulin receptor substrate. Insulin promotes neointimal hyperplasia and stimulates proliferation-related signaling pathways such as Akt-mTOR-P70S6K. Fig. 3 shows that the activation of the Akt-mTOR-P70S6K pathway was significantly increased after treatment with 1 µM of insulin for 15 mins. Compared to cells treated with insulin alone, cells treated with rosiglitazone before insulin stimulus demonstrated a significant inhibitory effect on the proteins upstream, Akt, and downstream, P70S6K, of mTOR. This suggests that rosiglitazone significantly inhibits the activation of the Akt-mTOR-P70S6K system.
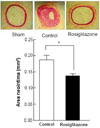
Neointima formation by rosiglitazone in balloon-injured OLETF rats
To test the role of rosiglitazone in neointima response after balloon injury, a daily dose of 3 mg/kg of rosiglitazone was administered to rats for 1 week before balloon injury and for 3 weeks after balloon injury. Withdrawal of the inflated balloon resulted in vascular injury, leading to neointima formation. Seven days after the injury, the inner vessel surface was covered with 1 or several layers of ovoid, irregular-shaped cells forming the neointima. In the present study, neointima formation in balloon-injured arteries was histologically confirmed. Fig. 4 shows that the density of neointima formation after balloon injury was much thicker than in sham-treated rats. However, no further neointimal growth was observed in OLEFT rats when treated with rosiglitazone after balloon injury. At the time of analysis, the neointima of OLEFT rats treated with rosiglitazone was thinner than that of controls.
Effects of PI3K or MEK inhibitors on Akt-mTOR-P70S6K in RAoSMCs treated with rosiglitazone
It was shown that rosiglitazone inhibits phosphorylation of the Akt-mTOR-P70S6K system in RAoSMCs stimulated with insulin. To determine whether factors upstream of the Akt-mTOR-P70S6K system regulate phosphorylation of this system, inhibitors of upstream molecules were added to cells prior to treatment with insulin. PI3K and MAPK are upstream molecules and related to the insulin-mediated signaling pathway, and insulin can activate PI3K through the phosphorylation of IRS1/2 or MAPK by Ras-GTP. It is also known that wortmannin is a PI3K inhibitor and U0126 inhibits MEK1/2 activation. Fig. 5 shows the effect of inhibition of the MEK/ERK and PI3K pathways on the effect of rosiglitazone treatment. The expression of p-P70S6K after rosiglitazone administration was assessed before and after treatment with MEK inhibitor U0126 (10 µM) and PI3K inhibitor wortmannin (10 µM). Inhibition of both MEK and PI3K resulted in partial reduction of p-P70S6K expression that was associated with additional inhibition, indicating that both the MAPK and PI3K pathways have an effect on the activation of the Akt-mTOR-P70S6K system.
DISCUSSION
Thiazolidinediones are insulin-sensitizing agents that are used to treat hyperglycemia by reduction of insulin resistance in type 2 diabetic patients.1 In addition to improvements in insulin sensitivity, studies have shown significant anti-inflammatory and anti-proliferative effects on the vascular tissues both in vitro and in vivo.2,12 However, the molecular mechanisms underlying the anti-proliferative effects have not yet been fully elucidated. In general, diabetes and hyperglycemia alone have not been suggested to be associated with the process of cell cycle regulation, however, insulin has been known to be a key player in protein synthesis, a factor important in cell proliferation. Insulin induces increased expression of translation initiation factors and phosphorylation of 4EBP-1 and S6K, critical cell cycle regulators downstream of mTOR. The mTOR is a key regulatory kinase that plays a major role in the mammalian cell cycle and is a member of a major pathway in the pathogenesis of neointimal hyperplasia and stent restenosis. Previous studies have revealed that mTOR signaling is important in cell and organism growth, cell cycle and proliferation, metabolism, and modulating gene transcription and transcriptional regulators. By inactivating P70S6K and eIF4E, rapamycin increases the activity of p27Kip1 and RB, subsequently resulting in inhibition of VSMC proliferation. Because mTOR itself is known to be activated by the PI3K-Akt pathway, we determined whether rosiglitazone inhibits VSMC proliferation through inhibition of the Akt-mTOR-P70S6K pathway and whether the Akt-mTOR-P70S6K pathway system is a specific major pathway for VSMC proliferation inhibition. This hypothesis was based on the fact that the Akt-mTOR-P70S6K pathway may be the upstream mechanism that increases the activity of p27Kip1 and RB with administration of PPAR-γ agonists. The results in the present study demonstrated that cells treated with rosiglitazone before insulin stimulus exhibited significant inhibition of p-mTOR, p-Akt, and p-P70S6K, indicating significant inhibition of the activation of the Akt-mTOR-P70S6K system. Namely, it has been known that insulin receptor signaling results in activation of PI3K and a variety of downstream effectors, including Akt, mTOR, and P70S6K, after insulin binds to insulin receptors located on the cell membrane. PI3K and a variety of downstream protein kinases, including Akt and p70S6K, represent 1 of the major signaling pathways mediating the metabolic effects of insulin.17,18 In the present study, the PI3K signaling pathway in primary cultured RAoSMCs was rapidly and markedly activated in response to insulin. Insulin sensitizers of the TZD class, which are ligands for the nuclear receptor PPAR γ, enhance insulin-mediated glucose uptake by ameliorating defective signaling from the insulin receptor to the PI3K pathway.19 Therefore, rosiglitazone blocks insulin-stimulated mTOR-related signaling in VSMCs.
However, there may also be a PPAR-γ-independent effect that inhibits the enzyme activity of this pathway, evidenced by the reduced levels of the phosphorylated forms of the pathway. Because the mTOR pathway members are activated by not only the PI3K-Akt pathway but also the mitogen-activated protein kinase pathway,20-22 the inhibitory effect of rosiglitazone on the Akt-mTOR-P70S6K pathway is regulated by both the PI3K-Akt and the mitogen-activated protein kinase pathway.
Previous studies have demonstrated that PPAR-γ activation reduces mitogen-induced VSMC growth by inhibition of Akt activity and increase of phosphorylated forms of 4E-BP1. It had been demonstrated that the sequence of events between the activation of Akt and increased phosphorylation of 4E-BP1 involves the activation of mTOR signaling. However, studies on the role of PPAR-γ agonists in modulating the activity of mTOR have not yet been done. This study demonstrated that the inhibition of the activity of the mTOR-P70S6K pathway may be an important mechanism involved in modulating the effects of various downstream signaling factors, such as the 4E-BP1 and CDK/CK complexes.
The interruption of blood flow caused by ligation of the common carotid artery induces migration and proliferation of SMCs from the media to the intima with concomitant infiltration and activation of circulating leukocytes, leading to neointima formation. OLETF rats characteristically begin to gain weight rapidly from 5 weeks of age, show insulin resistance at 8 - 9 weeks, and finally develop obese type 2 diabetes. The cumulative incidence of diabetes in OLETF male rats over age 23 weeks is 86%. In OLETF rats treated with rosiglitazone after balloon injury, the intensity of neointima formation and immunoreactivity of p-mTOR were markedly decreased.
In conclusion, rosiglitazone inhibits neointimal hyperplasia after carotid balloon injury because it blocks the Akt-mTOR-P70S6 kinase pathway, resulting in inhibition of VSMC proliferation. The inhibitory effect of rosiglitazone on the activity of the Akt-mTOR-P70S6K pathway may be mediated by the MAPK and PI3K pathways.




 XML Download
XML Download