

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Heparins (UFH)-like compounds, including low molecular weight heparin (LMWH), have been shown to have an important impact on the aggregation, degradation, and toxicity of β-amyloid peptide (Aβ),1,2 suggesting their potential roles in the treatment of Alzheimer's disease (AD). Since the compounds with high molecular weight (MW) can not penetrate the blood brain barrier (BBB), their therapeutic use is greatly diminished. Therefore, the compound with low MW may have more therapeutic value in the treatment of AD than the larger molecular substances. A LMWH of MW 4500 can bring about nuroprotection when administered both externally and intraneuronally, but UFH of M.W. 12,000 works only when injected into the cells,3 suggesting that the larger UFH molecule cannot pass through the cell membrane, while the smaller LMWH molecule does indeed enter the cell. Ultra low molecular weight heparin (ULMWH) is currently under development, and it exhibits a much smaller average MW and a much narrower distribution. It has been reported that ULMWH has a great ability to penetrate the BBB,4 indicating that it may have a much greater ability to modulate the functionality of the central nervous system. Our laboratory recently showed that ULMWH can protect brain from the ischemic injury in rats in vivo and its mechanism may be partly attributed to scavenging free radicals by antioxidant enzymes and modulating Ca2+ homeostasis.5 In order to deeply and extensively explore its neuroprotective effects, we performed lots of in vitro experiments and found that ULMWH could attenuate glutamate-induced cortical cell death (data not shown). As an extension of the research, we investigated the neuroprotective effects of ULMWH in the current study. The effects of ULMWH on glutamate- induced neurotoxicity, intracellular Ca2+ levels ([Ca2+]i), Bax/Bcl-2 expression and caspase-3 protease activation were investigated. Three basic questions were addressed: (1) whether ULMWH has protective effects on glutamate-induced neurotoxicity; (2) whether changes in ([Ca2+]i are induced by ULMWH or glutamate correlated with this protective function; (3) whether the protective effects of ULMWH are due to apoptosis/the apoptotic process.
MATERIALS AND METHODS
Materials
Ultra low molecular weight heparin (ULMWH) (mean MW= 2200) was sodium salt obtained from Institute of Biochemical and Biotechnological Drugs, School of Pharmacy, Shandong University. Pregnant Wistar rats were provided by Experimental Animal Center of Shandong University. Glutamate, Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum (FBS), poly-L-lysine, trypsin, B27 supplement, and neurobasal medium were all provided by Gibco (Logan, UT, USA). Hoechst 33258 was purchased from Sigma Chemical Company (St Louis, MO, USA). Cleaved caspase-3 was from Cell signaling (Beverly, MA, USA), and Bcl-2, Bax, and β-actin from Neomarker (Fremont, CA, USA). ECL was obtained from Amersham Pharmacia (Buckinghamshire, UK), Fura-2 AM was from Biotium (Hayward, CA, USA), and DNA extraction kit from Promega (Madison, WI, USA). All other chemicals used were of the highest grade available.
Cell cultures
Rat cortical cells were isolated from brain according to the procedures with slight modifications as described previously.6 Briefly, the cerebral cortice from E18 (embryonic day)-E19 Wistar rat was dissected under sterile conditions, triturated through a fire-polished pipettes, digested at 37℃ with 0.125% trypsin in d-Hank's solution for 15 min, and centrifuged at 310 g for 10 min. The resulting pellets were resuspended in Dulbecco's modified Eagle's medium (DMEM) and an aliquot was stained with 4% trypan blue for live cells count using a hemocytometer. The suspension was appropriately diluted with neurobasal medium containing 2% B27 supplement and 1% penicillin/streptomycin, and seeded in poly-L-lysine-coated 24, 96-well culture plates (Corning, Acton, MA, USA) or 75 cm2 Falcon Primaria flasks (Becton Dickinson, NJ, USA) at a density of (1 - 2) × 106 cells/mL in a humidified incubator with 5% CO2 overnight at 37℃, then the culture medium was completely replaced with fresh medium. During the subsequent 7 - 8 days culture, the cells were refed every other day by replacing only one third of the medium with fresh one. Few glial cells can be detected in the cortical cultures by using anti-rat neuronal specific enolase (NSE) and anti-glial fibrillary acidic protein (GFAP, unpublished observations).
Cell treatments
After cultured for 7 or 8 days, cortical cells in fresh medium were divided into four groups for different treatments: control cells (not treated with glutamate or ULMWH), 24 h treatment with 100 µmol/L glutamate, 24 h treatment with 1 mg/L ULMWH, and 24 h treatment with 1mg/L ULMWH followed by treatment with 100 µmol/L glutamate 24 h later. ULMWH was first dissolved in culture medium to give appropriated concentrations, and glutamate was diluted in phosphate buffered saline (PBS, pH 7.4).
Cell viability assay
The cultures were assessed for viability by MTT assay.7 In brief, 100 µL of MTT (0.5 mg/mL in PBS) was added to each well. After 4 - 6 h incubation at 37℃, 100 µLof DMSO was added and mixed well. Plates were agitated on a microtiter plate shaker to ensure a homogeneous solution, and optical densities (OD) at 570 nm were determined by using an automated spectrophotometric plate reader (Bio-Tek ELX808, Winooski, VT, USA).
Staining of chromatin using Hoechst 33258
Hoechst 33258 staining was used to reveal nuclear morphological changes in cultured cells.8 Cells were washed in PBS (× 3), fixed with 4% paraformaldehyde in PBS at room temperature for 30 min, rinsed with PBS (× 3), and then incubated with 0.1 mg/mL Hoechst 33258 for 30 min at room temperature. Morphological evaluation of nuclear condensation and fragmentation was performed using a fluorescent microscope (Olympus IX70-FL, Tokyo, Japan). The number of cells with apoptotic morphology was randomly assessed, and about 200 cells per field were counted.
DNA fragmentation
DNA fragmentation analysis was performed by the method reported by Wolozin et al.9 with minor modifications. Briefly, in order to extract DNA fragmented in the internucleosomal space, cells were cultured in 75 cm2 flasks. Approximately (1 - 2) × 107 cells from each experimental condition were harvested. The cells were washed with PBS and centrifuged for 5 min. The pellets were lysed in 10 mmol/L Tris-HCl (pH 7.5) containing 10 mmol/L EDTA and 0.2% Triton X-100. After centrifugation at 1600 g for 5 min, the supernatants were incubated with 1% SDS and 5mg/mL RNase A at 56℃ for 2 h and then digested with 2.5mg/mL proteinase K for at least 2 h at 37℃. After incubation and addition of 1mol/L ammonium acetate, the DNA was precipitated with ethanol and dissolved in gel loading buffer. The entire sample was then electrophoresed on 2% agarose gels containing 0.1 g/mL ethidium bromide, and DNA fragments were visualized under ultraviolet light.
Assessment of intracellular Ca2+ using fura-2/ AM
[Ca2+]i was measured microspectrofluorimetrically as previously described using the Ca2+-sensitive indicator fura-2/AM.10 Cells were incubated in 5 µmol/L fura-2/AM in DMSO for 45 min at 37℃, rinsed twice with d-Hanks to remove the dye, and then trypsinized, pelleted, and resuspended in d-Hanks containing 0.4% bovine serum albumin (BSA). The fluorescence ratio (R) was assessed using FDSS6000 at 340 nm and 380 nm wavelengths. The maximal fluorescence ratio (Rmax) was determined by adding Triton X-100 (final concentration, 0.1%), and subsequently minimal fluorescence ratio (Rmin) was determined by adding EGTA (final concentration, 5 mmol/L). The [Ca2+]i was then calculated according to the equation: [Ca2+]i= Kd (R-Rmin)/(Rmax-R), where Kd is the dissociation constant of fura-2/AM for Ca2+ and is assumed to be 224 nmol/L at 37℃.
Western blot
Cultured cortical cells were scraped into ice-cold homogenization buffer, containing 10 mmol/LTris-HCl (pH 7.4), 0.32 M sucrose, 1 mmol/L dithiothreitol, 1 mmol/L phenylmethylsulfonyl fluoride (PMSF), and 5 mmol/L EGTA, for 20 min at 4℃. After sonication, the cells were centrifugated at 4℃ (1000 g, 10 min) and the supernatants were used for the determination of protein concentrations using the Bradford assay. Samples were diluted (1 : 1) in sample buffer (62.5 mmol/L Tirs-HCl, pH6.8, 2% SDS, 5 mmol/L β-mercaptoethanol, and 10% glycerol) and boiled for 5 min.
The Western blot analysis was carried out as described previously.11 Briefly, 30 µg of protein from each sample were loaded onto a 12% sodium dodecyl sulfate-polyacrylamide gel (SDS-PAGE) for electrophoresis at 200 V for 30 min. Resolved protein samples were transferred from the gel to nylon membranes using an electrophoresis transfer apparatus, which was run at 12 V for 2 h. Membranes were blocked in 5% powered nonfat milk in Tris-buffered saline/Tween (TBST) solution (20 mmol/L Tris-HCl, pH 7.6, 0.1% Tween-20 in saline) overnight at 4℃. Membranes were then incubated for 1 h with a primary antibody diluted in blocking solution (1 : 100 for Bax, Bcl-2, and caspase-3). Membranes were then washed three times in TBST solution and incubated for 1 h with horseradish peroxidase-conjugated secondary antibody diluted in blocking solution (1 : 2000). Membranes were again washed three times, incubated with ECL detection reagent according to the manufacture's protocols, and then exposed to X-ray films. The amount of protein was quantified by densitometric analysis of the autoradiograms using a scanning laser densitometer. Protein levels of β-actin were determined to assure equal loading.
RESULTS
Effect of ULMWH on cell nuclear morphology and DNA fragmentation
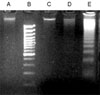
Nuclear shape and chromosomal structure can be visualized using Hoechst 33258 staining method.8 Cells are considered apoptotic if they show irregular, shrunk, and condensed nuclei with bright fluorescence. As seen in Fig. 1A and B, very few apoptotic cells were detected in the control (Fig. 1A) or ULMWH-treated group (Fig. 1B), however, significantly more apoptotic cells were observed in the glutamate-treated group (Fig. 1C), which was greatly reduced by ULMWH-pretreatment, as demonstrated in Fig. 1D. In order to confirm the glutamate-induced apoptotic morphology change and the effects of ULMWH pretreatment, DNA fragmentation study was then conducted on the sister cultures. As illustrated in Fig. 2, typically pronounced DNA ladder was observed in the cortical cells that were exposed to 100 µmol/L glutamate for 24 h, which was completely blocked by ULMWH pretreatment for 24 h and no DNA ladder was observed.
Quantification of the Hoechst 33258 stained apoptotic cells indicated that about 19.9% of control cells exhibited an apoptotic morphology, and there was no significant difference between control cells and ULMWH-treated cells. As compared with control cells, the percentage of apoptosis in glutamate-treated cells was 33.21%. However, the proportion decreased in ULMWH-pretreated cells, which had no significant difference from control cells (Fig. 3), thus confirming the qualitative observation.
Neuroprotective effect of ULMWH on cell viability
The viabilities of the treated cortical cells were studied using MTT assay. The optical densities (OD) of the cells were decreased by incubation with 100 µmol/L glutamate for 24 h, indicating that cells could be injured by excessive glutamate. Under this experimental condition, the decreased OD values could be attenuated by pretreatment with 1mg/L ULMWH before glutamate incubation (Fig. 4).
Effect of ULMWH on Bcl-2 family proteins expression
Since Bcl-2 family proteins play critical roles in apoptosis induced by a wide array of death signals, the effects of ULMWH on glutamate-induced changes of levels of Bcl-2 family proteins were studied using Western blot (Fig. 5A). Glutamate-treated cells showed highly elevated-Bax protein level with a concomitant down-regulated Bcl-2 content as compared with control cells. Such changes were reversed in the ULMWH-pretreated cells. There was no significant difference in the levels of Bax or Bcl-2 between ULMWH-treated cells and control cells (Fig. 5B). Errors in sample loading were controlled with β-actin.
Effect of ULMWH on glutamate-induced elevation of [Ca2+]i
As glutamate-mediated cell death may be correlated with increases of [Ca2+]i, we examined [Ca2+]i in the cells (Fig. 6). The level of [Ca2+]i in control cells was 137.68 nmol/L, whereas the glutamate-treated cells showed a significant rise of [Ca2+]i, which was partially inhibited by pretreatment with ULMWH. There was no significant difference between control and ULMWH-treated cells.
To investigate the [Ca2+]i-lowering pathways of ULMWH in cortical cells, the extracellular medium was changed to Ca2+-free medium containing 1 mmol/L EGTA (Fig. 7). However, glutamate-induced rise of [Ca2+]i was still observed, and the increase of [Ca2+]i was much smaller than that in normal culture medium. When 1 mg/L ULMWH was added to Ca2+-free medium prior to the application of glutamate, the glutamate-induced rise of [Ca2+]i was completely suppressed. Moreover, the magnitude of [Ca2+]i decrease by 1 mg/L ULMWH in normal culture medium was almost equal to that in Ca2+-free medium (normal culture medium: 20.76 ± 0.34 nmol/L; Ca2+-free: 19.9 ± 0.46 nmol/L, p > 0.05).
Effect of ULMWH on caspase-3 activation
As [Ca2+]i was elevated in glutamate-treated cells, its downstream protease, caspase-3, was assayed by Western blot analysis (Fig. 8). A significant 56% increase of caspase-3 protein was noted in glutamate-treated cells, as compared to that in control cells, and this increase was significantly attenuated in ULMWH-pretreated cells. There was no difference between control cells and ULMWH-treated cells or between control cells and ULMWH-pretreated cells. Again, uniform sample loading was assured using β-actin.
DISCUSSION
Glutamate is a major excitatory neurotransmitter working at a variety of excitatory synapses in the central nervous system. It plays important physiological roles in the cellular processes underlying synaptic plasticity, neuronal development and excitation via the activation of glutamate receptors.12,13 Of particular importance to all of these processes is the ability of the N-methyl-D-aspartate receptor channel to conduct Ca2+ ions, leading to a direct increase of intracellular Ca2+ through Ca2+ channel from the extracellular environment. Under physiological conditions, such as periods of patterned glutamatergic synaptic activity, the increase of intracellular Ca2+ can promote synaptic stabilization and strengntening.13-16 Under pathological conditions, such as prolonged exposure to high concentrations of glutamate, excessive amounts of intracellular Ca2+ are toxic, causing the so-called excitotoxic reaction.17,18 High concentrations of glutamate have been shown to induce neuronal damage and late cell loss in in vitro studies using cultured neurons.19,20 This is also observed in humans suffering from acute degenerative diseases such as strokes and traumatic brain injury, or chronic degenerative diseases including Parkinson's disease, AD and multiple sclerosis.21-23 Therefore, excessive glutamate accumulation in the extracellular space is believed to contribute primarily to injury to neurons, and high concentrations of glutamate are often used to mimic neuronal excitotoxity in vitro.
High concentrations of glutamate have been shown to produce excitotoxic damage in cultured neurons via Ca2+ entry throug receptor channels,24 and the glutamate-induced Ca2+ overload hypothesis has been widely accepted as the mechanism of neuronal injury in glutamate-induced excitotoxicity.25-27 Ca2+ overload should elicit a series of neuronal injury events, including degeneration of DNA, important proteins and phospholipids, and accumulation of radical oxygen species.28 In order to evaluate ULMWH modulation of changes in the level of intracellular Ca2+ induced by high concentrations of glutamate, we first assessed the intracellular free Ca2+ level in containing Ca2+ medium using fura-2/AM double wavelength fluorometry. The results showed that the intracellular Ca2+ concentrations were significantly increased by glutamate, but these increases were partially inhibited in the ULMWH-pretreated cells. Secondly, we observed changes in the level of intracellular Ca2+ after exposure to glutamate for 24 h in Ca2+ free medium. Intracellular Ca2+ concentrations were also increased by glutamate, but these increases were completely inhibited in ULMWH-pretreated cells. In addition, it was very interesting to note that the magnitude of [Ca2+]i decrease by ULMWH in medium containing Ca2+ was almost equal to that in Ca2+-free medium. This indicates that 24 h pretreatment of cortical cells with ULMWH can significantly decrease intracellular free Ca2+, which may be due to inhibition of the release of sequestered intracellular Ca2+. Although the way by which ULMWH inhibited the intracellular Ca2+ release wasn't directly investigated in the present study, UFH, the parent drug of ULMWH, has been proved to be an effective competitive antagonist of inositol-1, 4, 5-triphosphate (IP3) receptor, not only in isolated membrane preparations but also in living cells.29-31 Moreover, it has been observed that intraneuronal injection of UFH (MW 12,000) in vitro prevents glutamate-induced Ca2+ release from intracellular stores through blocking of IP3 receptors, thus interfering with events occurring in the ischemic cascade.3 ULMWH has average M.W. much smaller than that of LMWH and the same major internal structure, α-L-IdoA-2-SO4-β (I→4)-α-D-GIrNSO3-6-SO4, as UFH, suggesting that it can pass through the cell membrane and exert its intraneuronal effects. From these results, it can be inferred that ULMWH inhibited glutamate-induced intracellular Ca2+ release may be attributable to suppressing intracellular Ca2+ release from IP3- sensitive stores.
The elevation of the intracellular free Ca2+ level also mediates the activation of Ca2+ dependent enzymes,32 including lipases, phospholipases and proteases, which cause neurotoxicity by catalyzing the release of free fatty acids such as arachidonic acid from membrane phospholipids. Free fatty acids open membrane pores, thereby facilitating the influx of Ca2+ as well as releasing Ca2+ from internal stores. Intracellular free Ca2+ acts as a messenger to regulate growth, differentiation and cell death. Strong Ca2+ influx has been reported to be a signal for apoptosis.33 The crucial step of apoptosis is regulated by the expression of genes of the Bcl-2 family, which consists of both pro-apoptotic (e.g. Bax, Bcl-xs, Bad and Bak) and anti-apoptotic (e.g. Bcl-2, Bcl-xL, and mcl-1) molecules.34,35 Bcl-2 and Bax are the proto-type oncoproteins whose functions include the regulation of apoptosis.36,37 Bcl-2 protein is a cytosolic protein with a lipidanchoring domain, which allows it to target the nucleus and to inhibit apoptosis. Bax, a member of the Bcl-family, homodimerizes and forms heterodimers with Bcl-2 protein, thereby reducing its anti-apoptotic effect.37,38 Overexpression of Bax accelerates apoptotic death. In many cell types, Bcl-2 and Bax have been demonstrated to play a dominant role in the regulation of apoptosis.39 For example, Bcl-2 and Bax are considered to play a role in regulating the survival and maturation of chondrocytes in joint cartilage.40 In this study, we investigated the gene expression of the pro- and anti-apoptotic factors, Bax and Bcl-2, in cortical cells using immunoblot. The results showed that Bax: Bcl-2 ratio was increased by glutamate, but this increase was suppressed in ULMWH-pretreated cells. Bcl-2 is known to protect against apoptosis triggered by a wide range of factors.24,41,42 However, excess Bax favors apoptotic cell death induced by numerous stimuli.24 The data from this study strongly suggests that Bcl-2 family protein may be involved in the process of ULMWH to protect cortical cells from glutamate-induced apoptosis.
Outer stimuli can initiate apoptosis through the above mechanism and may converge on the caspase pathway to execute the final phase of the apoptotic process.43 The caspase family of proteases consists of at least 14 mammalian members that are constitutively expressed in almost all cell types as inactive proenzymes (zymogens) that become processed and activated in response to a variety of pro-apoptotic stimuli.44 Caspase-3 is a downstream member of the caspase cascade and acts as a central effector in the execution phase. When caspase-3 precursor protein CPP32 is activated by upstream signals such as the release of mitochondrial cytochrome c, triggered by Bax translocation to mitochondria,45 the active caspase-3 cleaves specific aspartate residues in proteins with various structural, housekeeping and regulatory functions.46 These proteolytic events can lead to cell apoptosis, and contribute to DNA fragmentation and nuclear morphologic changes. Thus, substances that can inhibit the activity of caspase-3 may protect cells from apoptosis.47,48 Since ULMWH markedly inhibited the caspase-3 activity in the glutamate-treated cells, it appears to have the neuroprotective capacity.
The spectrum of the anti-apoptotic potential of UFH and LMWH in various tissues and under various pathological situations49,50 is currently under scrutiny. In this study, our data clearly demonstrated that ULMWH can abrogate glutamate-induced apoptosis in primary cultured cortical neurons. Pretreatment of the neurons with ULMWH for 24 h can prevent the rise of [Ca2+]i triggered by glutamate via suppressing intracellular Ca2+ release from IP3-sensitive-internal stores, up-regulating the protein expression of the anti-apoptotic molecule Bcl-2, down-regulating the pro-apoptotic factor Bax, and preventing caspase-3 activation, all of which were involved in the mechanism of ULMWH conferred neuroprotection and can be viewed as a stepwise path to anti-apoptotic effect of ULMWH.







 XML Download
XML Download