

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The recombinant human growth hormone (rhGH) has been developed for adults with growth hormone deficiency (GHD), and its physiological and therapeutic effects are well documented in the literature.1-3 Notably, administration of rhGH is reported to antagonize the effects of insulin on lipid and glucose metabolism;4-9 in vivo studies demonstrate that rhGH administration can reduce insulin and glucose responses in rats.5,8,10,11
The mechanism responsible for the observed association between rhGH and insulin resistance is not well understood, but increased levels of free fatty acid (FFA) and reduced levels of GLUT-4 protein molecules have been implicated.12-17 Another plausible mechanism for insulin resistance is that elevated FFA levels can enhance the accumulation of intramuscular triglyceride (TG) content, which may contribute to insulin resistance in skeletal muscle. Some investigators have shown an inverse association between intramuscular TG content and insulin sensitivity.18,19 However, little is known about this association after rhGH treatment. Therefore, we investigated the effects of chronic in vitro rhGH administration and hypothesized that it will increase intramuscular TG content and reduce insulin sensitivity. The results from this study support that hypothesis.
MATERIALS AND METHODS
Animals
Fourteen 4-week-old Sprague-Dawley male rats (Samtaco Bio Korea, Inc; body weight 128.5 ± 3 g) were housed one per cage in a 12 : 12 light-dark cycle at 24℃, and were given Harlan rat chow (Harlan Teklad) with tap water ad libitum. After 7 days of adaptation to the laboratory environment, the rats were randomly assigned to two groups: 1) saline injection group (CON, n = 7) and 2) rhGH injection group (GH, n = 7). The GH group received rhGH (Eutropin®, LG inc, Gyeonggi, Korea) by subcutaneous injections (130 µg·kg-1·day-1, 6 days·week-1) for 4 weeks, while the CON group received saline injections as equivalent volume as the GH group. All protocols were approved by the Kyungpook National University Animal Care and Use Committee.
Oral glucose tolerance test (OGTT)
After 4 weeks of treatment and following a 12 hours fast, the rats were given 50% aqueous glucose solution (1 g·kg-1 body weight) using a stomach tube. The rats were placed in acrylic restrainers on a heating pad, and approximately 0.5 mL of blood was taken from the tail immediately prior to glucose administration, and at 30 and 60 minutes after administration. To determine plasma insulin levels, blood samples were placed in microcentrifuge tubes (Cole-parmer international, Vernon hills, IL, USA) containing 30 µL Heparin (Choongwae Pharma. Co., Jeonbuk, Korea), and centrifuged at 10,000 g for 20 minutes. The plasma portion was removed and stored at - 80℃ until further analysis. Insulin resistance was determined under submaximal insulin concentrations during muscle incubation.
Tissue sampling
One week after the OGTT, the rats were fasted for 8 hours and then anesthetized by intraperitoneal injection with sodium pentobarbital (6.5 mg·kg-1). The epitrochlearis muscles were surgically isolated and removed to assess glucose transport rate in muscle,20 while the liver and plantaris muscle were excised to assay the TG content. Blood samples were drawn from an abdominal aorta to measure plasma glucose, insulin, total cholesterol, triglyceride (TG), high-density lipoprotein cholesterol (HDL-C), and FFA levels.
Determination of glucose transport in skeletal muscle
Glucose transport activity was measured using 3-MG (3-O-[3H]-methylglucose). After dissection, muscles were rinsed briefly in 25-mL flasks containing 3 mL of KHB (Krebs-Henseleit buffer, 0.1% BSA) and then transferred to recovery vials with a recovery medium (32 mM mannitol, 8 mM glucose). Following this 30 minutes recovery period, the muscles were transferred to pre-incubation vials, containing KHB as a preincubation medium (8 mM glucose, 32 mM mannitol, and 0.1% BSA), and then incubated for 20 minutes. After 20 minutes of pre-incubation, the muscles were rinsed with fresh KHB medium-supplement (40 mM mannitol) and transferred to final incubation vials. The final incubation medium contained 3-O-[3H]-methylglucose (2.2 µCi·mmol-1) and [14C] mannitol (0.2 µCi·mmol-1) in the presence or absence of a submaximal (1,000 µIU·mL-1) dose of insulin. Throughout the incubation process, samples were maintained at 35℃ during the recovery and pre-incubation periods; at 30℃ during the rinse; at final incubation in a shaking water bath (120 cycles·min-1); and at gassed with a 95% O2 and 5% CO2 mixture. The muscles were then blotted, clamp-frozen, and processed for determination of intracellular 3-MG accumulation and extracellular space. To determine glucose transport rate, muscle samples were analyzed in a liquid scintillation counter (Beckman Instruments, Inc, Fullerton, CA, USA) by setting the channels at simultaneous 3H and 14C readings.21
Determination of GLUT-4 in skeletal muscle
Portions of gasrtrocnemius muscle were homogenized for 15 seconds in ice-cold HES buffer (20 mM HEPES, 1 mM EDTA, and 250 mM sucrose), using a motor driven homogenizer (Art-Miccra D-8 Model, Art Labortechnik, Müllhein, Germany). Sample homogenates and standards were diluted by 1 : 2 with 2x Laemmli sample buffer (S3401, Sigma-Aldrich, St. Louis, MO, USA) and incubated for 20 minutes. Muscle homogenates, which contained 50 µg of protein, were then subjected to SDS-polyacrylamide-gel-electrophoresis under reducing conditions of a 10% resolving gel. Resolved proteins were transferred to a nitrocellulose membrane (BioRad, Hercules, CA, USA) and blocked for 60 minutes with 5% non-fat milk. Membranes were incubated with GLUT-4 antiserum (Santa Cruz Biotechology, Santa cruz, CA, USA), diluted with 1 : 10,000 in a T-TBS/5% dry milk, for 90 minutes. Membrane were washed with T-TBS and incubated with secondary antibody (ZYMED Labolatories; at a dilution of 1 : 5000 with T-TBS/1% non-fat milk) for 60 minutes at room temperature, and washed with T-TBS. The GLUT-4 protein was visualized by Hyperfilm (Eastman Kodak, Rochester, NY, USA) using the Western blot luminal reagent (Santa Cruz Biotechology, Santa cruz, CA, USA).
Determination of ceramide in skeletal muscle
Plantaris muscles were homogenized in buffer (0.25 M sucrose, 25 mM KC1, 50 mM Tris, 0.5 mM EDTA; pH7.4). After homogenization, lipid was extracted by adding a chloroform, methanol, and buffer water mixture maintained at the ratio of 0.5:1.0:0.4 and 1.0:1.0:0.9 (v/v/v) before and after dilution, respectively. Lipid extraction and ceramide determination were carried out according to the guidelines described by Bielawaka et al.22
TG contents in muscle and liver
Liver samples were homogenized in 50 mM potassium phosphate and 1 mM EDTA (pH 7.4) with 1 : 15 dilution, while plantaris muscle samples were homogenized as 1 : 20 dilution. Lipid was determined by following guidelines described by Burton and Anderson.23 Homongenized samples were centrifuged at 1,000 g for 10 minutes, and the supernatant was removed by adding 0.5 mL of 2-propanol to prevent any evaporation of TG. The TG content was measured using an enzymatic method (Elitech, Sees, France) and quantified in the skeletal muscle of both groups.
Biochemical measurements
Plasma glucose concentrations were determined by a glucose analyzer (Model YSI-23A, Yellow Springs Instruments, Yellow Springs, OH, USA). Plasma insulin and leptin concentrations were measured via radioimmunoassay (Linco Research Inc., St. Louis, MO, USA) by using a double antibody procedure.24 Plasma TC, TG, and HDL-C levels were measured by an enzymatic method (Elitech, Sees, France). Plasma FFA was determined by an acyl-CoA oxidase-based colorimetric kit.25
Statistical analysis
Independent t tests were used to compare mean differences for FFA levels, GLUT-4 contents, TG contents, and glucose transport rates between the GH and CON groups. Two-way ANOVA with repeated measures were used to determine plasma glucose and insulin responses across group by time factor. The sphericity assumption was justified using Hyunh-Feldt Epsilon (ε) test. Pearson correlation with simple regression was used to investigate the association between TG content and glucose transport rate. All statistical procedures were performed by SPSS (ver. 14) with a significance level at 0.05.
RESULTS
Growth rate
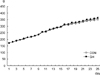
There was no statistical difference in growth rate between the GH and CON groups (Fig. 1).
Biochemical characteristics
Biochemical characteristics after 4 weeks of rhGH treatments are presented in Table 1. The GH group (0.83 ± 0.07 mmol·L-1) had higher FFA values than the CON group did (0.58 ± 0.05 mmol·L-1, p < 0.05). There were no statistical differences in TC, TG, and HDL-C values between the 2 groups.
Oral glucose tolerance test
As shown in Fig. 2, the GH group had higher glucose levels than the CON group at basal time (GH 132.6 ± 5.0 vs CON 98.9 ± 4.5 mg·dL-1, p < 0.05) and at 60 minutes (GH 182.0 ± 13.2 vs CON 148.7.90 ± 6.1 mg·dL-1, p < 0.05) as compared with the CON group, respectively. The GH group also had higher insulin levels at basal time (GH 1.95 ± 0.36 vs CON 0.98 ± 0.07 µU·mL-1, p < 0.05), at 30 minutes (GH 4.53 ± 0.42 vs CON 2.54 ± 0.35 µU·mL-1, p < 0.05), and at 60 minutes (GH 3.62 ± 0.21 vs CON 1.81 ± 0.21 µU·mL-1, p < 0.05) as compared with the CON group, respectively.
GLUT-4 contents in muscle
There was no statistical difference in GLUT-4 contents between the GH (RG: 37.7 ± 2.0%, WG: 21.3 ± 1.5%) and CON (RG: 36.6 ± 2.3%, WG: 22.3 ± 1.6%) groups (Fig. 3).
TG contents in muscle and liver
Fig. 4 shows that the GH group (1.56 ± 0.12 µmol·g-1) had greater TG content in the plantaris muscle compared with the CON group (0.79 ± 0.08 µmol·g-1, p < 0.05). The GH group also had significantly higher TG content in the liver (9.29 ± 1.46 µmol·g-1) than with the CON group (5.12 ± 0.59 µmol·g-1, p < 0.05).
Ceramide contents in muscle
Fig. 5 shows that the GH group (83.8 ± 2.1 pmol·mg-1) had greater ceramide content in the skeletal muscle than with the CON group (75.7 ± 2.7 pmol·mg-1, p < 0.05).
Glucose transport rate in muscle
As shown in Fig. 6, the GH group had a lower glucose transport rate under submaximal insulin concentrations (2.36 ± 0.18 µmol·mL-1·hr-1) than the CON group (3.26 ± 0.26 µmoL·mL-1·hr-1, p < 0.05). However, there was no statistical difference between these 2 groups under non-insulin conditions.
Triglyceride content and glucose transport rate
Fig. 7 shows the association between glucose transport rate and muscle TG content. There was a significant inverse association between muscle TG content and glucose transport rate (r = - 0.67, p < 0.01).
DISCUSSION
In the present study, we observed a decrease in plasma glucose and insulin responses during OGTT and glucose transport rate during muscle incubation under submaximal insulin concentration in rhGH injected rats (130 µg·kg-1·day-1 for 4 weeks). Also, we observerd that prolonged exposure to rhGH elevated intramuscular TG and ceramide levels in rats. In addition, plasma FFA and hepatic TG content were also elevated when rhGH was administered. Thus, the major finding of this study is that the decreased insulin sensitivity due to chronic rhGH administration would partly result from an elevated intramuscular ceramide content which is deriveded from the elevated muscle TG content.
It has been reported that there was an inverse relationship between insulin sensitivity and intramuscular TG stroage,19,26 similar to the results of the present study. However, elevated intramuscular TG storage in itself did not reduce muscle insulin sensitivity, but rather acts as a source for lipid derivatives, such as diacylgycerol, long chain fatty acid acyl-CoA or ceramides, that directly affect insulin signalling and, thus, insulin sensitivity.27-29 In previous studies,27 these was a possibility that ceramide might inhibit insulin signalling at the level of protein kinase B (PKB)/Akt.30 Therefore, we hypothesized that the decreased glucose transport due to rhGH injection might partly result from elevations of intramuscular TG storage and ceramide content. In the current investigation, we observed that rhGH administation significantly elevated muscle TG storage and ceramide content. Thus, intramuscular ceramide elevation due to rhGH injection may partly contribute to the development of insulin resistance
It has also been postulated that FFA levels due to rhGH treatment are responsible for insulin resistance.31,32 The elevated FFA levels demonstrated here is consistent with previous reports.16,33 Many previous studies reported that an increase in circulating FFA is a key factor for the development of insulin resistance in insulin sensitive tissues such as adipocytes and skeletal muscle.13,34,35 Increased level of FFA by rhGH treatment has been suggested to be related to insulin resistance.14,16,36 In vitro studies have indicated that the lipolytic actions of rhGH may involve stimulation of gene expression after binding to the rhGH receptor and subsequent activation of JAK2 tyrosine kinase, adenyl cyclase, and stimulation of cAMP production, triggering the hormone-sensitive lipase by increasing circulating FFA.37-39 According to the glucose-FFA cycle postulated by Randle et al.,40 increased FFA concentrations may decrease the uptake of glucose in skeletal muscle. Skeletal muscle is responsible for 70 - 80% of whole body insulin-stimulated glucose uptake and is, therefore, considered to be the most important site of insulin resistance.13 The mechanism by which raised plasma FFA concentrations inhibit insulinstimulated glucose uptake in muscle may involve the insulin-signaling pathway responsible for GLUT4 translocation, including IRS-1 phosphorylation, PI3K activity, or glycogen synthase activity.41-44 In addition, an inverse correlation between muscle lipid content and insulin sensitivity has been demonstrated in humans by muscle biopsy,19 computed tomography45 and magnetic resonance spectroscopy.26 TG stored within the muscle fiber has been implicated as a casual factor in the insulin resistance-FFA relationship, and the postulated mechanism involves intramyocellular accumulation of diacylglycerol, activation of protein kinase C, or myoplasmic long-chain fatty acyl CoA.46,47
We also observed that GLUT-4 protein levels were similar between the GH and CON groups. GLUT-4 is the major glucose transporter protein in skeletal muscle, and the muscle GLUT-4 protein levels are significantly related to the rate of glucose transport during muscle incubation.48 Therefore, any decrease in the rate of glucose transport in rhGH-treated rats would seem to require a reduction of GLUT-4 levels in skeletal muscle. However, our data showed no reduction of muscle GLUT-4 protein levels in the rhGH-treated rats, and suggest that the deflect in the signalling mechanism occurred prior to the glucose translocation process. This result is consistent with the previous reports that muscle GLUT-4 protein levels were not altered in rhGH-treated rats,10,41 and supports the concept that alterations of muscle GLUT-4 protein levels are not responsible for the reduction of insulin-stimulated glucose disposal in rhGH-treated rats.
It is plausible that rhGH-treated rats develop insulin resistance in skeletal muscle due to elevated intramuscular TG storage and ceramide content. Elevation of circulating FFA levels due to rhGH treatment enhances intramuscular TG content, and high levels of muscle TG and ceramide content are responsible for insulin sensitivity in skeletal muscle. Our data also showed that reduced glucose transport rate in the rhGH-treated rats occurred with no change in GLUT-4 content, and was directly related to the TG content of the muscle. A strength of this study is that, to our knowledge, this is the first study to specifically investigate the association between muscle TG content and glucose transport rate after the rhGH treatment. Our study is also well designed to eliminate all possible confounding factors using a randomized control design. Further studies are needed to validate our findings across different murines as well as in humans.
In conclusion, we found that rhGH treatment is directly associated with insulin resistance in rats. The causative factor for insulin resistance is that rhGH treatment elevates TG content and ceramide in muscle, and an elevated TG and ceramide contents are inversely associated with insulin sensitivity in rats.







 XML Download
XML Download