

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
DAA is usually an isolated aortic arch anomaly and is rarely (7% to 17%) associated with other congenital heart diseases.1,2 DAA may manifest as respiratory distress and feeding problems early in life. The cardinal features of respiratory distress and feeding problems, which are not uncommonly encountered in patients with isolated DAA, can be masked by the overwhelming cyanosis or congestive heart failure is frequently seen in patients with DAA associated with other major cardiovascular malformations. In this retrospective study, the clinical, radiographic, echocardiographic, angiographic, and magnetic resonance imaging (MRI) characteristics of isolated and complex DAA and its differentiation from the d-TGA, HTA, type I TA, and APW were reported to familiarize pediatric practitioners with these congenital heart diseases.
MATERIALS AND METHODS
From July 1, 1994 to December 31, 2006, a total of 6 patients (4 male and 2 female, aged 16 days to 6.5 years) with DAA and 38 patients with CTM, including d-TGA (n = 28), HTA (n = 3), type I TA (n = 4), and APW (n = 3) were included in this retrospective survey. Patients with incomplete vascular rings, or right aortic arch with left ligamentum were excluded.3 To support the diagnosis of DAA, d-TGA, HTA, type I TA, or APW, we monitored the patients with chart recordings (n = 44), plain chest films (n = 44), plus at least 2 of the following study modalities: two-dimensional echocardiography with Doppler (Acuson 128XP/10c, Mountain View, CA, USA) (n = 44), cardiac catheterization with angiography (n = 44), surgery (n = 42), MRI (n = 2), barium esophagography (n = 6), and chromosome analysis (n = 2).
Echocardiograms and angiocardiograms of patients with the DAA were compared with those of the patients with d-TGA, HTA, type I TA, and APW. The absolute bifurcation distance, from semilunar aortic valve to pulmonary bifurcation (d-TGA, HTA, and APW), or from truncal valve to aortopulmonary bifurcation (type I TA) was measured directly by the echocardiographic calipers or indirectly by the angiographic calipers after calibration of the catheter. This absolute distance was corrected for body surface area (BSA) in terms of mm/m2. Magnetic resonance imaging studies were performed on 2 patients with DAA to delineate the associatedintracardiac lesions. To confirm diagnosis of the 22q11 deletion syndrome, cytogenetic analysis and fluorescence in situ hybridization (FISH) studies were performed on 2 patients presenting with the clinical stigmata of DiGeorge sequences. One patient had DAA and tetralogy of Fallot, and the other patient had APW, type B aortic interruption, patent ductus arteriosus, and ventricular septal defects.
RESULTS
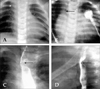
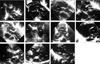
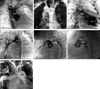
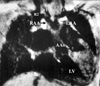
All 4 male and 2 female patients, aged between 16 days and 6.5 years, had a remarkable history of respiratory distress and postprandial choking since birth. Two patients had isolated DAA, while four had associated complex cardiovascular malformations. Recurrent infections of the lower respiratory tract were noted in 5 patients with DAA. A 16-day-old neonate with 22q11 deletion syndrome did not manifest recurrent lung infection due to the early diagnosis and repair of the DAA and the tetralogy of Fallot. Remarkable cyanosis was found in 3 patients since birth, which were caused by intracardiac anomalies rather than DAA. One of these patients had tetralogy of Fallot and the 22q11 deletion syndrome, one had tetralogy of Fallot and absent pulmonary valve syndrome, and one had tricuspid atresia and pulmonary atresia. Congestive heart failure was noted in one patient with large ventricular septal defects. Inspiratory stridor was noted in 4 patients and expiratory wheezing lingered in the patient with the tetralogy of Fallot and the absent pulmonary valve syndrome, a syndrome which is usually associated with respiratory problems. Dysphagia was found in 3 children. Two out of the 6 patients tolerated respiratory and feeding problems from birth to 14 and 15.5 years of age, respectively. Plain chest films showed a suspicious contour of the right aortic arch in 2 patients (Fig. 1A) and deviation of the trachea in 1 patient (Fig. 1B). Barium esophagograms, which were performed in 5 patients, showed an indentation of the esophagus (Figs. 1C and D). DAA was appreciated before cardiac catheterization by two-dimensional echocardiography, in which suprasternal and subcostal views of the left ventricular outflow tract offered diagnostic windows for the DAA (Fig. 2A). As for CTM, subcostal and parasternal views can always determine the cause to be either d-TGA (Figs. 2B and K), HTA (Figs. 2C, F and I), type I TA (Figs. 2D, H, and J), or APW (Figs. 2E and G). The ascending aortography showed a bifurcated, or Y-shaped double aortic arch (Figs. 3A and B), with a dominant right arch, and a smaller left arch in 5 patients. One patient had balanced aortic arches. Left ventriculography of the d-TGA patients (Fig. 3C), and ascending aortography of the HTA, (Fig. 3D), type I TA (Figs. 3E and F), and APW (Fig. 3G) patients demonstrated that the bifurcations originated from one great artery, either the main pulmonary artery or the ascending aorta, which was committed to the left ventricle. Magnetic resonance imaging studies, which were performed in 2 patients with complex DAA, showed a bifurcated or Y-shaped double aortic arch, with a dominant right arch and a smaller left arch (Fig. 4). All but 2 patients underwent surgery at the time of presentation and diagnosis. Four (2 with isolated DAA, 1 complicated with VSD, and 1 with tetralogy of Fallot and 22q11 deletion syndrome) out of the six patients with DAA survived the surgical division of the smaller left aortic arch. The former 3 surviving patients were repaired by a one-stage surgery, and the latter surviving patient was repaired by a two-stage procedure. Surgical interventions were declined by the parents of the remaining two patients, one with the absent pulmonary valve syndrome and one with tricuspid atresia as well as pulmonary atresia. The two patients, who did not undergo surgery, could tolerate mild cyanosis, exertional dyspnea, and dysphagia in their daily activities. At the time of this report, these two patients continue to survive without surgical correction and have oxygen saturations between 80% and 85%.
All patients with d-TGA (n=28) presented with remarkable cyanosis, while patients with HTA (n = 3), type I TA (n = 4), and APW (n = 3) manifested overt congestive heart failure. Among the 3 patients with HTA, 2 had isolated lesions and one had associated Berry syndrome. All 4 patients with type I TA had associated large ventricular septal defects. In 3 patients with APW, one patient had isolated APW, one patient had APW associated with the 22q11 deletion syndrome (DiGeorge sequences, type B aortic interruption, patent ductus arteriosus, and ventricular septal defect), and one patient had APW associated with the Berry syndrome (type A aortic interruption, patent ductus arteriosus, ventricular septal defect, and hemitruncus arteriosus). Patients with DAA had a much longer bifurcation distance and a larger bifurcation distance/BSA ratio (bifurcation distance 20-66 mm, median 45 mm; bifurcation distance/BSA ratio 72-114 mm/m2, median 97 mm/m2; patient age, 16 days to 6.5 years old) than those measured in the patients with CTM (bifurcation distance 5-10 mm, median 8 mm; bifurcation distance/BSA ratio 24-55 mm/m2, median 42 mm/m2; within 7 days of life), including the patients with d-TGA (10 mm/0.181 m2 = 55 mm/m2), HTA (9 mm/0.195 m2 = 46 mm/m2), type I TA (5 mm/0.208 m2 = 24 mm/m2), and APW (8 mm/0.189 m2 = 42 mm/m2).
The clinical and echocardiographic characteristics of DAA and the characteristics that differentiate DAA from CTM are summarized in Table 1.
DISCUSSION
DAA is the most frequently encountered vascular ring malformation that inevitably completely encircles the trachea and esophagus.1 The DAA, together with the right aortic arch with left ligamentum, is defined anatomically as the complete vascular ring.3 DAA is usually found as an isolated cardiovascular malformation. Associated cardiac anomalies occurred only in 8 patients (7%) in a large study of 113 patients with DAA1 and appeared in 14 patients (17%) in another study of 81 patients with DAA.2 Among these associated cardiac anomalies, ventricular septal defects rank first, atrial septal defects follow at distant second, while patent ductus arteriosus, tetralogy of Fallot, d-TGA, and many others follow far behind.2-14
The cardinal manifestations of respiratory distress and feeding problems, which are commonly encountered in patients with isolated DAA, can be masked by the overwhelming features of cyanosis or congestive heart failure that is often seen in patients with DAA complicated by other intracardiac anomalies. For example, in patients that have DAA intricated with tetralogy of Fallot,4,5 d-TGA,6-8 tricuspid atresia, and pulmonary atresia, they will also have overt cyanosis rather than respiratory distress, postprandial choking, and aspiration pneumonia. Patients with large ventricular septal defects, truncus arteriosus,9 patent ductus arteriosus,10 coarctation of the aortic arch,11-13 and supravalvular mitral stenosis with subvalvular aortic stenosis, and ventricular septal defects14 may present with discernible congestive heart failure rather than with the above major features of respiratory and gastroesophageal symptoms. The clinical or diagnostic spectrum of a vascular ring may range from severely compromised respiratory and gastroesophageal symptoms in newborns with complete vascular rings (DAA or right aortic arch with left ligamentum) to freedom from symptoms in adults with incomplete rings due to an aberrant subclavian artery. In every newborn or child with stridor, wheezing, and dysphagia, the presence of a complete vascular ring should be carefully identified. In asymptomatic adults, the diagnosis of an incomplete vascular ring is usually made by chance.
Many radiological modalities can provide important clues toward a diagnosis of DAA. First of all, plain chest radiographs may show a deviation or compression of the trachea, or contour of a right arch, especially in the scenario of an infant presenting with respiratory and gastroesophageal symptoms. Barium esophagography has been advocated as a valuable investigation for patients with suspected vascular rings, because it can be safely and rapidly performed to diagnose DAA with high sensitivity. Barium esophagograms were once considered the single most important or the primary method of study in evaluating patients with vascular rings.15,16 However, this method is no longer considered a sufficient evaluation of the patient before proceeding with treatment by vascular ring repair.1 This trend in imaging diagnosis for vascular rings may change because of the increasing ease and safety provided by computed tomography (CT) and MRI and the ability of these methods to improve patient outcome through their precise ability to plan operative procedures.1 Compared to CTs and MRIs, barium esophagograms cannot provide a clear surgical "road map" that can generate a precise preoperative strategy,1 especially in patients with typical vascular ring formations that necessitate using a right thoracotomy for surgical treatment.17-19 For the evaluation of patients with vascular rings, CT and MRI could be used as valid alternative methods to barium esophagograms and angiographies.20,21 This dramatic shift from barium esophagograms and angiographies to CTs and MRIs in the preoperative evaluation strategy of 209 patients with complete vascular rings (113 patients with DAA and 96 with right aortic arch and left ligamentum) may be stemmed from the following: 1) referrals were generated by otolaryngology instead of pediatric cardiology, 2) a significant number of patients with right aortic arches with left ligamentum, 3) the unusual anatomy of a right aortic arch with a left ligamentum, retroesophageal left subclavian artery, and large Kommerell diverticulum, and 4) the inability of the echocardiographers to identify useful anatomic details.1 However, these updated options shall not lead us to leave the echocardiography and barium esophagograms in the niche.
Two-dimensional echocardiography can usually substantiate a diagnosis of DAA. Furthermore, echocardiography is very useful for identifying associated congenital heart diseases. Subcostal22 and suprasternal23,24 views of the two-dimensional echocardiography may offer diagnostic windows for pediatric practitioners or cardiologists to assess patients with DAA. The subcostal view of the left ventricular outflow tract can be used to visualize aortic bifurcation, which was 40 mm and 45 mm above the aortic valve in 2 infants. This bifurcation generates a Y-shaped, centripetal, and embracing morphology.22 We cannot overemphasize that the subcostal approach is practical only in infants younger than 12 months old, especially in neonates. A major drawback of the subcostal approach is that the area of interest is confined to far-field imaging, which can be given a reduced resolution in larger patients. The suprasternal notch approach can overcome this problem by imaging the area of interest in the near field. By a suprasternal notch long-axis/sagittal plane (with leftward obliquity) and a 30 degrees counterclockwise rotation of the transducer (with rightward obliquity), we can visualize a left arch and a right arch, respectively. In each oblique plane, we can see only 2 head and neck arteries, i.e., carotid (1st branch) and subclavian (2nd branch) arteries, arising sequentially and ipsilaterally from each corresponding arch.25 Then, a suprasternal notch short-axis/coronal plane can be used to track down the possible culprit to be a DAA by using one scanning plane. We shall not jump to display both arches in one suprasternal notch short-axis/coronal plane without identifying the branching pattern of each arch in the suprasternal notch long-axis or sagittal plane a priori. Jumping to the conclusion of a DAA diagnosis by only performing a suprasternal notch short-axis/coronal view is risky, due to the large number of false positive images which are caused by a "phantom" right pulmonary artery aliasing as a vascular ring. In the hands of experienced investigators, transthoracic echocardiography permits an accurate imaging of DAA and any associated congenital heart diseases as well as clear identification of the sidedness of the aortic arch. The major problem resides in its limited ability to image clearly the atretic aortic arch structures and a ligamentum arteriosum. Nonetheless, echocardiography remains a powerful, useful, and non-invasive tool that is capable of evaluating patients with suspected DAA.
From the subcostal view of the left ventricular outflow tract, we can differentiate the DAA from the d-TGA, HTA, type I TA, and APW by visualizing a centripetal morphology of the aortic bifurcation and by measuring the bifurcation distance. The aorta bifurcates at a strikingly longer distance beyond the aortic valve in patients with DAA. Such characteristic morphology can be easily differentiated from the pulmonary arterial bifurcation morphology seen in d-TGA cases22 and the anomalous take-off morphology of the aortic pulmonary artery, namely, hemitruncus arteriosus, truncus arteriosus, and APW. With the provisos of patients' symptoms and esophageal indentations, the subcostal and suprasternal echocardiographic views offer diagnostic windows for DAA.9,22,23 By the subcostal approach, we have found that these two bifurcations "embrace" each other centripetally in patients with DAA. From the embryogenetic viewpoints, the bifurcation distance from the semilunar valves to the pulmonary or aorticopulmonary bifurcations should be shorter than the bifurcation distance observed in DAA cases.22,26 All these differences can be derived from embryogenesis due to branchial arch defects in DAA cases and conotruncal septation defects in patients with other malformations.26 By approaching from the suprasternal notch, echocardiography can identify the dominant arch, the nature of each arch, and its branches.22-25 Parasternal long and/or short-axis views and subcostal left ventricular outflow tract views are helpful in excluding DAA from a diagnosis, since early bifurcation is seen in one of the great arteries in patients with d-TGA, HTA, type I HA, and APW.
Although cardiac catheterization with angiography can provide a diagnosis of DAA and a differential diagnosis of rare types of aortic arch anomalies, pulmonary slings, and ductal slings,27-31 catheterization is neither routinely required nor clinically feasible for smaller patients with severe cardiopulmonary distress. Instead, CTs and MRIs can be performed non-invasively and safely, especially in patients compromised with severe cardiac or pulmonary failure. CTs and MRIs, which can image the atretic aortic segments, are of great value for preoperative planning.1
In patients with isolated DAA, surgical division of the smaller aortic arch is indicated. However, preoperative recognition of associated anomalies is important, especially in cases with coarctation or atresia of one or both arches, associated major congenital heart diseases, tracheomalacia, and bronchomalacia. On the one hand, surgical division of one aortic arch, without preoperative recognition of coarctation or atresia of the other arch, will make clinical care demanding and can be life threatening. On the other hand, in patients associated with other congenital heart diseases, e.g., tetralogy of Fallot, d-TGA, HTA, HA, and APW, which may dominate the clinical manifestations with cyanosis and heart failure, the symptoms of DAA may be too subtle to appreciate. Finally, a preoperative bronchoscopy can be used to rule out associated tracheomalacia or bronchomalacia and is important to the patient's short- and long-term prognosis.1 Thus, echocardiography, angiography, bronchoscopy, CT, and MRI can be selectively employed to evaluate the detailed anatomy of the aortic arches and airways in patients with DAA.
In conclusion, visualization of the contour of a right aortic arch with or without tracheal deviation on plain chest radiographs of infants presenting with respiratory and gastroesophageal symptoms may lead to an early suspicion of DAA. Altogether, barium esophagography, echocardiography, angiography, bronchoscopy, CT, and MRI can be applied individually to evaluate the detailed pathology of the aortic arches, airways, and the associated congenital heart diseases as well as permit a detailed planning of successful surgical correction in patients with DAA.

 XML Download
XML Download