

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Behçet's disease (BD) is a chronic, relapsing, multi-systemic, inflammatory process with the clinical features of mucocutaneous lesions, and ocular, vascular, articular, gastrointestinal, neurologic, urogenital, pulmonary, and cardiac involvement. The etiopathogenesis of the disease remains unknown. Although several immunological abnormalities have been demonstrated in patients with BD, the exact mechanism of the inflammatory changes occurring also remains to be elucidated. The most likely hypothesis seems to be that of an autoimmune reaction set off by infectious agents such as human herpes virus 1 or Streptococcus species in genetically predisposed individuals, and its basic pathologic process is vasculitis.1-5
Because of the lack of a universally recognized pathognomonic laboratory test, the diagnosis of BD is primarily based on clinical criteria. Several sets of diagnostic criteria have been used, and all of these criteria rely heavily on mucocutaneous manifestations, in particular oral ulcers (OU), genital ulcers (GU), cutaneous vasculitic lesions and pathergy reaction. The International Study Group for Behçet's Disease6 developed, in 1990, new, internationally agreed,7 diagnostic criteria which depend on the presence of recurrent OU, relapsing at least three times in a 12-month period, plus any two of recurrent GU, typical eye lesions, typical cutaneous lesions or a positive pathergy test (Table 1).
BD usually occurs around the third decade of life and has a chronic course with unpredictable exacerbations and remissions. The disease is a universal disorder, however, with a very variable prevalence of 80-370 per 100,000 in Turkey, with 80 per 100,000 in the European part of Turkey, 150 per 100,000 in the Ankara region and 370 per 100,000 in the North-Eastern part of Turkey, 14-20 per 100,000 along the Silk route and perhaps within 100 kilometers on either side but very low elsewhere in Asia, and 30 per 100,000 in Northern Japan (Hokkaido region) but only 1 per 100,000 in Southern Japan (Kyushu region). In contrast, 0.1-7.5 per 100,000 are involved in Europe and the USA.8,9 The sex distribution is roughly equal. In contrast to early reports of male predominance from Turkey10 and Japan,11 the male-to-female ratio has drastically decreased in the last 20 years to currently reach an equal rate8,12-14 with the sole exception of the Arab countries where the male predominance persists.8 This probably results from the fact that in strict Muslim societies, male doctors cannot examine women and women doctors are rare. In some Western societies, women are more likely to present as patients in doctors' offices, regardless of diagnoses. Male sex, a younger age of onset and HLA B51 positivity in BD are associated with more severe disease.8 In a multivariate analysis of a mixed German- Turkish sample, the combination of male age and vascular lesions (uveitis, thrombophlebitis, erythema nodosum, etc.) as an onset sign exhibited a significantly worse prognosis that any other group, whereas in the group of patients with OU as onset sign, male HLA-B51 positive patients showed a most common development of eye disease (our own unpublished data). Eye disease, papulopustular lesions (PPL) and thrombophlebitis are also more common among males. The disease rarely develops before puberty or after the age of 50, and it generally runs a milder course in women. Clinical manifestations of BD, with the exception of eye symptoms, tend to improve with time.15,16 Serious complications such as central nervous system involvement and sight threatening eye disease are rarely observed at late onset, especially in cases of onset at 40 years of age or more.17 Recent studies18 suggest that besides considerable morbidity, the disease confers an increased mortality, mainly because of central nervous system, pulmonary and large vessel involvement and bowel perforation. There is evidence that a lethal outcome is often due to delayed diagnosis and treatment.
Mucocutaneous lesions constitute the hallmark of the disease. The high frequencies of OU, GU and cutaneous lesions at any stage in the course of the disease, or as onset signs, confirm the importance of these clinical features for the diagnosis. OU (92-100%), GU (57-93%), cutaneous lesions (38-99%) and ocular lesions (29-100%), together with arthropathy (16-84%), generally mild, mostly arthralgias, and when arthritis, often monoarticular, are the most frequent clinical features of the disease in all countries.8,19
Erythema nodosum (15-78%) and PPL (28-96%) are the most commonly observed cutaneous lesions.8,19,20 OU represent the onset feature of the disease in the majority of the patients worldwide (47-86%). GU (0-18%) and skin lesions, especially erythema nodosum (0-19%) can also occur as onset lesions.8,19 The skin pathergy test is a unique feature of the disease and the test positivity varies (6-71%) widely in different populations.8
Our recent study19 showed that the disease is often diagnosed with a delay of several years after the appearance of the onset sign. Duration between the time point of fulfillment of diagnostic criteria and the diagnosis was found to be 2.83 ± 2.3 years. In the majority of patients, mucocutaneous lesions, especially OU, appear before potentially severe organ involvement occurs. In our study, OU were the most commonly observed onset manifestation (85%). The duration between OU and the fulfillment of diagnostic criteria was calculated to be 3.77 ± 4.43 years. Therefore, familiarity with the cutaneous spectrum is imperative for prompt recognition of the disease.
Oral ulcers
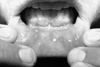
OU are characterized by recurrent and painful ulcerations of the oral mucosa. Patients may have single or multiple ulcers which often subside spontaneously after a couple of weeks, but then recur at intervals from days to months. They are identical to conventional aphthae or recurrent aphthous stomatitis (RAS) in appearance, but they tend to be more frequent and occur in crops21 (Fig. 1). Increased number of ulcers (more than six at the same time), concurrent variation in size from that of herpetiform to major aphthous, diffuse erythematous surrounds and involvement of soft palate and oropharynx have been suggested to differentiate OU of BD from conventional RAS.22 Bang et al.23 assessed the prognosis of RAS by performing prospective evaluations of 67 patients who had only a history of RAS. Thirty-five patients (52.2%) developed overt manifestations of BD at an average of 7.7 years after the onset of RAS. The frequency of recurrence was 9.8 times per year in progressive cases.
OU recurring at least three times over a 12-month period are the key to the diagnosis of BD.6,24 These lesions are the most frequent onset sign and the most commonly observed manifestation in all countries. Because of their high incidence at any stage of the disease, OU are crucial to the diagnosis. It was shown that only 1-3% of patients with BD analyzed by the International Study Group lacked recurrent OU.6 Therefore, recurrent OU should be regarded as one of the diagnostic manifestations for BD, and patients should be followed at regular intervals, using clinical criteria for evolution into BD, especially in the Mediterranean and Far East regions.
OU usually occur on the nonkeratinized oral mucosa. The most common sites of the lesions are the mucous membranes of the lips, the buccal mucosa, the floor of the mouth, the soft palate and the undersurface of the tongue. Keratinized oral mucosa, hard palate, gums and dorsal tongue are rarely involved (Fig. 2). The tonsilar fauces and pharynx can also be involved. The lesions start as an erythematous, circular, slightly raised area evolving into an oval or round ulcer within 48 hours. The lesions are painful with rolled or overhanging borders and a grayish yellow necrotic base. An erythematous rim surrounds the ulcer. OU often last 1 to 4 weeks, and local trauma can induce new mucosal lesions (mucosal pathergy equivalent). OU can cause remarkable pain and may interfere with eating, speaking and swallowing.24-27
They can be classified as minor, major or herpetiform on the basis of ulcer size and number.
1. Minor ulcers are defined as isolated or multiple shallow ulcers, frequently 3-6mm in diameter (< 10mm). Minor ulcers are often found on the nonkeratinized mobile mucosa of the lips, cheeks and floor of the mouth, sulci or underside of the tongue. Rarely they are observed on the gingiva, palate or dorsum of the tongue. The number of lesions varies and can be more than 6 simultaneously. They usually heal in 1-2 weeks without scarring. Clearly, the term minor is for classification purposes only; no lesions which present for medical attention are minor to the patient.
2. Major ulcers are morphologically similar to minor ulcers, but larger (> 10mm), deeper, and more painful than minor ulcers. These ulcers may involve any region of the mouth. They last longer and heal with scarring and tissue loss. Generally, few ulcers occur at one time and they heal slowly over 10-40 days.
3. Herpetiform ulcers are numerous (up to 100), shallow, small-pinpoint (1-2mm in diameter) ulcers occurring in coalescing clusters. They can be present at any oral site and can heal with scarring. Sometimes herpetiform ulcers increase in size and coalesce to form large ragged ulcers.
Conditions to consider in the differential diagnosis of OU are those that produce recurrent ulcers on the oral mucosa. Herpes simplex, herpetiform, minor or major forms of RAS, some cases of erythema multiforme, fixed drug eruption, and acatalasemia (Takahara's disease) have a recurrent course. Herpes simplex presents with grouped, small, shallow ulcers. Herpetiform lesions can also be seen in the course of conventional RAS and BD. A Tzanck smear should be performed in all cases with such lesions. If multinucleated acantholytic cells are seen, the diagnosis is herpes simplex. However, herpes simplex virus-PCR is more reliable. Target lesions, especially on the acral and periorificial areas, indicate erythema multiforme, whereas a nummular erythematous base and a history of recurrence of erosions at the same site after each intake of the causative drug, fixed drug eruption and vesicles on the hands and feet, indicate hand-foot- mouth disease. In cases beginning in childhood and involving the alveolar area, the hydrogen peroxide test should be performed to exclude acatalasemia. This disease sometimes destroys the jaw. When the physician excludes all these causes, the diagnosis of RAS is established. RAS may be a part of BD, besides MAGIC syndrome, Reiter's syndrome and Sweet's syndrome, or may be secondary to anemia due to a deficiency of iron, vitamin B12 or folic acid or to inflammatory bowel diseases (Crohn's disease and ulcerative colitis). OU are one of the American Rheumatism Association's criteria for the diagnosis of systemic lupus erythematosus. Almost half of such lupus erythematosus patients have OU with irregular and slit-like appearance. Lesions are usually located on the palate and often heal with a scar. The major form of RAS is also called Sutton's disease. It is characterized by well-defined, larger (1cm in diameter or larger), deeply punched-out, painful ulcers, which heal with scarring, and may be associated with cyclic neutropenia besides the causes mentioned for RAS above. In patients with HIV infection, although the incidence of aphthous ulcers is not increased, the lesions are more numerous and deeper, so that they interfere with nourishment and lead to severe weight loss.28
Complex aphthosis, first described by Jorizzo et al.,29 or malignant aphthosis, first described by Tourraine (1941), are the terms used to describe patients who have almost constant, multiple (> 3) OU or recurrent OU and GU without systemic manifestations of BD. Differentiation of BD from the complex (malignant) aphthosis can be problematic since the initial clinical presentation of BD is often confined to OU and GU. In our opinion, patients with complex aphthosis must be monitored at intervals using clinical criteria for evolution into BD.
Genital ulcers
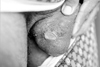
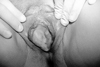
GU are the second main symptom encountered. The lesions are present in varying proportions, ranging from 57 to 93%.8 They are similar in appearance and course to OU, but may not recur as often and can have a scarring tendency. Therefore, a careful examination should also include a search for genital scars, even if there are no signs of active ulcers at the time. GU are usually deeper than the OU and their appearance can be preceded by a tender nodule. They are usually painful or occasionally asymptomatic, especially in female patients.24-27
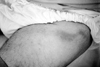
The scrotum is the most frequently involved (90%) site in males (Fig. 3). Ulcers can also be observed on the shaft and glans penis. In females, the ulcers most commonly occur on the labia but the vaginal mucosa and rarely the cervix can also be affected (Fig. 4). Vaginal ulcers often cause a discharge. Deep ulcers may scar and those in the vagina may be complicated by bladder or urethral fistulae. Vulval ulcerations occasionally led to labial destruction. GU may occur in both sexes in the groin, perineal and perianal area.30,31
In the differential diagnosis, conditions presenting with recurrent GU should be considered in the first place.
In herpes simplex infection, lesions arise as multiple, painful, small, grouped vesicles on an erythematous base and recur at the same location each time. The genitalia are also a preferred site for both erythema multiforme and fixed drug eruption. Besides conditions presenting with recurrent GU, when rapidly evolving erosion or ulcer/s are present without showing any clue for blistering both in history and physical examination, sexually transmitted diseases, mainly syphilis, chancroid, lymphogranuloma venereum and HIV infection should be investigated with appropriate laboratory methods.
Cutaneous lesions
Cutaneous lesions are another important feature of the disease and have been described by the International Study Group for Behçet's Disease as a major criterion for the diagnosis.6 The cutaneous lesions of the disease vary and mainly include erythema nodosum-like lesions, PPL, superficial thrombophlebitis, extragenital ulceration, reactivity of the skin to needle prick or injection (pathergy reaction) and other cutaneous vasculitic lesions.
Papulopustular lesions
PPL, the most common skin lesions, are cutaneous, sterile, folliculitis - or acne -like lesions on an erythematous base which appear as a papule and in the course of 24-48 hours become pustule20,32 (Fig. 5). Although four of the five new international criteria for the diagnosis of BD relate to mucocutaneous lesions, disagreement exists as to the exact nature of the cutaneous lesions and the inclusion of cutaneous follicular or acneiform lesions as a major criterion. Many authors33 believe that these lesions should not be included, since acneiform and folliculitis-like lesions are nonspecific and clinically, may not be differentiated from ordinary acne, particularly in adolescents. In a randomized and controlled study,20 we counted PPL, including acneiform and folliculitis- like lesions, in a blind protocol according to the 7 different anatomic locations. The frequency of PPL in patients with BD was 96%, and the most common location was the trunk, followed by the extremities, whereas in the control group the frequency was 89% and the most common location was the face (the frequency was 100% and 86.1% in acne and non-acne patients, respectively). Our study suggests that PPL is very sensitive but not specific when acneiform or follicle-based lesions were also included. To differentiate acne lesions from the PPL of BD can be very important, especially in those patients for whom the diagnosis depends on this criterion. In a recent study, on the basis of the above studies,20,33 we excluded follicle-based acneiform lesions and those lesions over the face since the clinical differentiation from acne vulgaris is nearly impossible at this anatomic location. Instead we investigated whether PPL can be a useful tool for the diagnosis of BD when non-follicular lesions over the trunk or extremities were selected, and were correlated with histologic and/or immunofluorescence study. Our results confirm that the selection of non-follicular lesions and combination with histopathologic and/or immunofluorescence studies increase the specificity of these lesions to establish the diagnosis.34 On the contrary, the definitive histological diagnosis from the cutaneous lesions is also often difficult without clinical information. Therefore, the clinician should consider correlating the diagnosis of PPL for BD by histological examination and only vessel-based histopathology (leukocytoclastic vasculitis or a neutrophilic vascular reaction) should be included as fulfilling a diagnostic criterion.
Erythema nodosum-like lesions
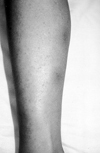
Erythema nodosum-like lesions of BD occur in about one-third of all patients. The lesions are mostly seen in females and are present 15-78% of patients.8 They occur mainly on the lower extremities (Fig. 6), but can also be seen on the face, neck and buttocks. The lesions do not ulcerate and generally resolve within 2 to 3 weeks, in pigmented ethnic groups with residual pigmentation, but recurrence is common. Although erythema nodosum-like lesions of BD are clinically similar to those of erythema nodosum secondary to other systemic causes, they differ from that condition with regard to their microscopic features. Vasculitis or vascular reaction-unlike classical erythema nodosum lesions- are the main features of these lesions, as they are in other cutaneous lesions of BD.35,36
Thrombophlebitis
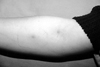
The prevalence of vascular involvement in BD has been reported to be 7.7-60% in different patient populations. Koc et al.37 have shown that the venous system is the major affected site (88%), and subcutaneous thrombophlebitis is, indeed, the most frequent type of venous involvement (47.3%). Superficial thrombophlebitis is frequently confused with erythema nodosum. The patients usually present with erythematous, tender, subcutaneous nodules arranged in a linear fashion (Fig. 7). The subcutaneous venules of the extremities, especially in male patients, tend to develop thrombosis leading to sclerosis. The small vein can be palpated as a string-like hardening of the subcutaneous tissue with reddening of the overlying skin. The location of nodules shows a tendency to change from day to day because multiple segments of the vein might be involved, resembling migrating obliterative thrombophlebitis. Besides subcutaneous veins, thrombotic process in BD may also involve the deeper veins of the brain, liver or lungs.38,39
Extragenital ulcerations
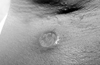
Extragenital ulcerations are a different type of cutaneous lesions and occur in about 3% of patients. Clinically, the lesions resemble aphthous lesions of the disease. They are recurrent and usually heal with scarring. The lesions have been reported on various locations such as the legs, axillae (Fig. 8), breast, neck, interdigital skin of the foot, inguinal region and neck.40 Extragenital ulcerations are common in children with the disease,41,42 are among the most characteristic and specific findings of the disease, and are rarely observed in other skin diseases.
Pathergy test
The skin pathergy test is a non-specific, skin hyperreactivity test, induced by needle prick or intracutaneous injection, and is associated with papule or pustule on an erythematous base, similar to the spontaneously occurring PPL of BD. Test positivity is defined as at least a papule observed at the needle-prick site 48 hours after application of a sterile needle that penetrated to the corium of an avascular site on the forearm (Fig. 9).43 The test positivity varies between geographical areas (6-71%), and has been reported to be especially high in Japan and the Mediterranean sea countries. About 60-70% of patients with BD in these regions are said to evidence skin hypersensitivity to needle prick. However, test positivity is uncommon in individuals living in the Western World, which reduces its diagnostic value in these countries.8 The Pathergy reaction is more strongly positive in males.44-46
The mechanism of the reaction remains obscure. Several authors incriminate cell-mediated immunity in the pathogenesis of this response. It was shown that the surgical cleansing of the skin surface before the application of the needle reduced the test positivity. Some substances, bacteria or skin products, eliminated by the surgical cleansing, might play a role in the development of pathergy test.46
The skin pathergy test is an outstanding feature of the disease, and has long been included among the criteria for diagnosis in many centers. The diagnostic reliability has been questioned and some authors have proposed that the histopathological and immunopathological, rather than clinical, assessment of pathergy sites are more reliable diagnostic adjuncts.45
Other cutaneous vasculitic lesions
Other cutaneous vasculitic lesions include Sweet's syndrome-like,47,48 pyoderma gangrenosum-like,49 erythema multiforme-like lesions,50 palpable purpura,51 subungual infarctions, hemorrhagic bullae,52 furuncles, and abscesses.24
The spectrum of the cutaneous lesions has been expanded by case reports with new cutaneous lesions such as pernio-like cutaneous lesions,53 polyarteritis-like lesions,54 and acral purpuric papulonodular lesions55 (Fig. 10). These relatively rare cutaneous findings may be part of this peculiar disease, but could also be coincidental. Their significance is still a matter of controversy and further observations are needed to decide on the clinical relevance. Therefore, Schreiner and Jorizzo25 postulated that only those lesions documented by neutrophilic vascular reactions or leukocytoclastic vasculitis should be included as cutaneous lesions of BD.
Histopathology
Histological assessment of cutaneous lesions from patients with BD describe primarily vasculitis and thrombosis.56 The histological features of early cutaneous lesions are leukocytoclastic vasculitis with karyorrhexis of neutrophils, extravasation of erythrocytes, and fibrinoid necrosis of postcapillary venules, or a neutrophilic vascular reaction with no fibrinoid necrosis surrounded by a neutrophilic infiltrate, nuclear dust, and extravasation of erythrocytes. Histological assessment of late lesions and autopsy data describe primarily lymphocytic perivasculitis. Histopathological examination of pathergy reaction from patients with BD also shows leukocytoclastic vasculitis, or a neutrophilic vascular reaction similar to that seen in Sweet's syndrome.24,33,45,57,58
These histopathological findings correlate with the theory that immune complex-mediated vasculitis is involved in the pathogenesis of BD, and that the vascular endothelium is primarily involved in the development of the lesions.59-61 Many authors believe that OU, GU, pustular cutaneous lesions, erythema nodosum-like lesions, Sweet's syndrome-like lesions, pyoderma gangrenosum-like lesions, and pathergy reaction are all variants of immune complex-mediated, neutrophil-induced, vessel-based lesions.58,62,63
Taken together, it seems likely that clinical lesions are still the most important criteria to establish diagnosis and, therefore, that clinical knowledge of the mucocutaneous lesions for a definitive histological diagnosis is required.
Treatment of mucocutaneous lesions
No standard therapy has yet been established for the disease. However, a wide spectrum of therapeutic agents have been used in the treatment of mucocutaneous lesions of the disease, with varying success. However, none of them cure the disease, and some are associated with significant adverse effects. The choice of treatment is generally based on the establishment of individual clinical manifestation/s. In mild forms of the mucocutaneous disease, initial therapeutic measures consist of mild diet64 and avoidance of hard, spicy or salty nutrients and chemicals. Topical treatment of OU includes:
- caustic solutions (silver nitrate 1-2%, tinctura myrrhae 5-10% w/v, H2O2 0.5%, methyl violet 0.5%) 1-2x/d,
- topical antiseptic and anti-inflammatory preparations (amlexanox 5% in oral paste65 hexetidine 1%, chlorhexidine 1-2% mouthwash solutions, benzydamine,66 camomile extracts), as well as tetracycline mouthwash (as glycerine solution 250mg/5ml glycerine) for 2min, 4-6x/d67 (caution: pregnancy),
- topical corticosteroids (triamcinolone mucosal ointment, dexamethasone mucosal paste, betamethasone pastilles) 4x/d or during the night (ointment/paste) or intrafocal infiltrations with triamcinolone suspension, 0.1-0.5mL per lesion,
- topical anaesthetics (lidocaine 2-5%, mepivacaine 1.5%, tetracaine 0.5-1% gels or mucosal ointments) 2-3x/d (caution: allergy),
- topical sucralfate (suspension, 1g/5mL) 4x/d, 3-month duration as mouthwash (has been shown to reduce the frequency, healing time and pain of OU in a randomized, double-blind and placebo controlled study21)
- topical aminosalicylic acid (5% cream) 3x/d (has been shown to reduce the duration of aphthous lesions and the pain intensity68).
For the topical treatment of GU and cutaneous lesions, corticosteroid and antiseptic creams can be applied for a short period of time (7 days). Painful GU can be managed by topical anaesthetics in cream. Topical sucralfate reduces the healing duration and pain of GU.21 Corticosteroid injections (triamcinolone 0.1-0.5mL/lesion) can be focally applied in recalcitrant ulcerations.
In severe forms of the mucocutaneous type of the disease, additional systemic treatment is required. The following agents have proven beneficial:
- Corticosteroids (prednisolone, initial dose 30-60mg/d p.o. for at least 4 weeks) can be administered as monotherapy or in combination with colchicine (1-2mg/d p.o.), Dapsone (100-150mg/d p.o.), IFN-α(3-12 Mill. IU/3x week s.c.) or azathioprine (initial dose 100 mg/d p.o.).69
- Non-steroidal anti-inflammatory drugs, like indomethacin (100mg/d p.o. over 3 months) can be effective occasionally on the mucocutaneous lesions.70
- Pentoxifylline (300mg 1-3x/d p.o.) and oxypentifylline (400mg 3x/d p.o.) treatment for 4 weeks induced a remission of OU in patients with BD and RAS, respectively, in two thirds of patients in open studies.71,72 However, recurrences occurred in all patients after discontinuation of treatment. Pentoxifylline (600 mg/d p.o.) has also been described as an alternative treatment for mucocutaneous and ocular lesions in a few patients with BD. The compound decreases superoxide production by neutrophils.
- Colchicine (0.5-2mg/d p.o.) can be used as a second-line alternative. A recent randomized, double-blind and placebo controlled study73 has shown that colchicine reduces the occurrence of GU, erythema nodosum, and arthritis among women. It inhibits the enhanced chemotactic activity of neutrophils. Oligozoospermia and gastrointestinal complaints are known to be adverse effects of colchicine (caution: pregnancy).
- Dapsone (100-150mg/d p.o.) also inhibits the enhanced chemotactic activity of neutrophils and can be used as an alternative compound to colchicine.74 Quick relapses have been observed after discontinuation of dapsone treatment. Intermittent treatment with ascorbic acid (vitamin C; 500mg/d) is advisable to prevent increased methaemoglobin serum levels.
- Interferon-α has been successfully used in the treatment of BD. Its immunomodulatory effect, ability to augment the decreased activity of the patient's natural killer cells, capacity to inhibit neovascular proliferation, and antiviral activity have been suggested to explain its action in BD.27,75 Currently interferon-α-2a was shown to markedly inhibit IL-8 synthesis and secretion from endothelial cells which may be a key event in its antiinflammatory activity.76 In a review of the relevant literature, Zouboulis and Orfanos75 concluded that a majority of patients showed a worthwhile improvement in mucocutaneous lesions, arthritis and ocular manifestations. A minimum two-month treatment is likely to be necessary to increase the effectiveness, and the disease generally relapses upon discontinuation. In a recent randomized, double-blind and placebo controlled study,27 Alpsoy et al. have shown that interferon-α 2a treatment at a dose of 6 Mill. I.U./3x week s.c., for 3 months, is an effective alternative, particularly for the management of mucocutaneous lesions of BD, and its effect decreases gradually after the cessation of treatment. The primary side-effects of IFN-α 2a therapy are flu-like symptoms (fever, chills, headache, fatigue, myalgia etc) that start a few hours after the initiation of the therapy, and continue less than a day. Nausea, vomiting, anorexia, diarrhea, loss of weight, hematologic changes, and transient raising of hepatic transaminases are seen less frequently.
-Azathioprine (2.5mg/kg body weight/d p.o.) has been found to be an effective choice in OU and GU besides ocular inflammation and arthritis in a randomized, double-blind and placebo controlled study.77
-Cyclosporin A (3mg/kg body weight/d p.o.) is capable of markedly ameliorating mucocutaneous lesions; however, it should be reserved for the most severe cases because of its significant long-term adverse effects.78
-Methotrexate (7.5-20mg/1x week p.o. over 4 weeks) is able to induce an improvement of a severe mucocutaneous involvement (caution: pregnancy, lactation, severe bone marrow depression, liver dysfunction, acute infections, kidney insufficiency, and mucositis and similar ulceration by its own effect).79
-Thalidomide (100-300mg/d p.o., optimal dose 100mg/d in the evening for 2 months) has recently been approved for the treatment of male and sterilised as well as post-menopausal women with BD in the USA.80 The drug was shown to selectively inhibit tumour necrosis factor (TNF)-αsynthesis by monocytes. In a randomized, double blind, placebo controlled study with 63 patients,81 a remission of OU, GU and PPL was detected in 24% of the patients over 8 weeks. During the 6-month treatment, 30% of the patients remained free of lesions. Discontinuation of the treatment results in OU and GU recurrence; therefore a maintenance treatment with 50mg/d to 50mg twice a week is required. Peripheral neuropathy with acral paraesthesia was found clinically in 6%, and electrophysiologically in 22%, of the patients who received thalidomide 100-300mg/d over 6 months. Central nervous system signs with sleepiness and headaches as well as xerostomia and constipation can occur.
-Recent trials of anti-TNF82-84 for BD have shown encouraging results in the treatment of recalcitrant OU and GU besides gastrointestinal symptoms. At the time of writing, two such compounds have shown favorable results in preliminary tests: infliximab, administered intravenously, and etanercept.
In conclusion, clinical lesions are still the most important criteria to establish the diagnosis of BD, and it is important to bear in mind the possibility of BD when confronted with recurrent OU, GU and/or other respected cutaneous lesions. As a high incidence of vital organ involvement, as well as an increased mortality, especially in young male patients, has been recorded in patients with BD, continuous surveillance is warranted in these cases. In this respect, patient-based organizations that are allied with cognizant physicians should be encouraged. With the recent advances in understanding the pathogenesis of the underlying disease, and the availability of a wide spectrum of therapeutic agents, alleviation of most symptoms, or control of the disease and, perhaps, modification of its course are now possible.



 XML Download
XML Download