

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Sarcoma of the kidney is a rare condition. Even though leiomyosarcoma is the most common renal sarcoma, transitional cell carcinoma is the most common neoplasm of the renal pelvis.1,2 Renal leiomyosarcomas have no pathognomic finding that would allow differential diagnosis. In the majority of cases, renal leiomyosarcoma is diagnosed through pathologic examination after surgical resection of the renal mass.3 Moreover, a combined congenital anomaly in the kidney can make it more difficult to diagnose the renal mass. This is a case report of a leiomyosarcoma that originated from the blind end of a bifid renal pelvis.
CASE REPORT
A 42-year-old female patient was admitted because of a cystic mass on her left kidney. The cystic mass was incidentally identified during a routine health evaluation. There was no relevant past history. Results of the physical examination were unremarkable. Laboratory data, such as blood chemistry and urine analysis that included urine cytology, showed normal levels.
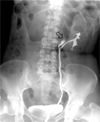
The computed tomography (CT) demonstrated a homogeneous cystic mass at the upper pole of the left kidney (Fig. 1A). An irregularly shaped solid mass of 5 × 3cm was adjacent to the cystic mass on the renal hilar area. The solid mass showed irregular enhancement in the arterial phase (Fig. 1B). To determine whether the solid mass was intrarenal or extrarenal, we used a retrograde pyelography that revealed a bifid left renal pelvis. We used radiological contrast media to visualize the pelvis and calices of the lower moiety. However, we did not visualize the upper moiety calyx (Fig. 2). Selective urine cytology of the upper moiety calyx was negative for malignancy. The patient underwent nephrectomy in order to remove the renal mass. A 5 × 3-cm protruding mass in the left renal hilum was found intraoperatively. En-bloc resection of the left kidney with the masses was performed.
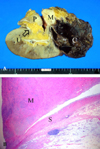
According to the pathologic examination, the kidney was 15.0 × 8.7 × 6.0cm and weighed 174g. The cystic lesion at the upper pole of the left kidney was a simple cyst. The renal pelvis of the upper moiety was blind-ended and was filled with a gray-white solid mass. The mass was 5 × 3cm and well-marginated. Moreover, portions of the mass showed a grayish mucoid appearance emerging (Fig. 3A). Tumor necrosis was not identified in the solid mass. In the low power microscopic field, the mass arose from the smooth muscle layer of the renal pelvis (Fig. 3B). Furthermore, the mass showed a few atypias of the nucleus, a few mitotic figures, and positive staining for smooth muscle-specific actin and for desmin (Fig. 4). S-100 and HMB-45 staining were negative. In the high-power microscopic field, the invasion of the atypic cells into the normal smooth muscle tissue was identified. Consequently, the solid mass was diagnosed as leiomyosarcoma which arose from the upper moiety of the bifid renal pelvis, combined with a large simple renal cyst at the upper pole of ipsilateral kidney.
DISCUSSION
Leiomyosarcomas of the pelvis of the kidney are extremely rare. To the best of our knowledge, leiomyosarcoma of the renal pelvis has never been reported in Korea to date. Only 10 cases have been described worldwide. Although information about the clinical evaluation of previously reported cases is limited, none of them were associated with other kidney anomalies.
Leiomyosarcoma usually has an irregular shape, and CT or MR imaging often reveals a heterogeneously enhanced, soft tissue mass without calcification or a fat component. Sonography or angiography has been useful in previous studies for defining the vascular structure and invasion when a mass lesion was found.3 The physical features of these masses, thus, make it difficult to preoperatively diagnose renal leiomyosarcoma purely based upon physical and radiological examinations.4 Furthermore, the combined anomaly in the kidney makes its diagnosis more difficult. Ureteroscopic inspection and biopsy and fine needle aspiration cytology could, therefore, be useful as a new method of preoperative diagnosis.5
Renal leiomyosarcoma composes 50-60% of all renal sarcomas and is, thus, the most common form among renal sarcomas.1 Renal leiomyosarcoma arises from the renal capsule or the smooth muscle fibers of the renal pelvis or vasculature.2 Microscopically, the classic spindle-shaped cells with hyperchromatic nuclei and varying numbers of mitoses are diagnostic for leiomyosarcoma. However, it is difficult to differentiate leiomyosarcoma from the sarcomatous form of renal cell carcinoma, because sarcomatous renal cell carcinoma also has spindle-shaped atypical cells. An immunofluorescent intermediate filament stain can differentiate leiomyosarcoma from renal cell carcinoma.1 Because the mass arises from the smooth muscle layer of the renal pelvis, no further examination for differential diagnosis is required.
Because renal sarcoma is a very rare condition, no effective treatment has yet been established. Complete surgical resection with at least a 3-cm safety margin is the treatment of choice3 but is rarely feasible due to the invasion of adjacent structures by the tumor. The most important prognostic factor is the tumor-free resection margin. Large mass size and frequent metastasis to adjacent organs is found in most cases at the time of diagnosis, and, consequently, the prognosis of most leiomyosarcomas is poor.3 Although radiotherapy, chemotherapy, or hormonal treatment is considered as adjuvant treatment, these treatments do not diminish tumor recurrence or improve the survival rate.3
Analysis of the 66 cases of renal leiomyosarcoma by Niceta et al.6 found higher morbidity in females than in males, rapid growth and high metastatic tendency, and both local and distant recurrence of renal leiomyosarcoma. Moreover, almost all patients died within two years despite radical nephrectomy.6 The Mayo Clinic reported that the survival for 14 of 15 patients with renal leiomyosarcoma ranged from 4 months to 5.5 years.6
Miyajima et al.7 suggested that the age and sex of the patient, tumor depth, tumor size, frequency of mitotic figures, degree of necrosis, shape of the nucleus, and AJCC tumor stage are the important prognostic factors. They additionally stated that the tumor size and the AJCC tumor stage are the most important of these prognostic factors. The five-year survival rate for tumors less than 5cm is 77.44%, whereas the survival rate drops to 41.86% if the tumor is larger than 5cm. The AJCC soft tissue sarcoma tumor stage of our case is T2aN0M0 GI and stage IB.7,8 Even though tumor size is larger than 5cm in diameter, because the cancer stage and grade is low, this patient would have a five-year expected survival of about 53% according to AJCC on cancer staging.
Janice et al.9 insisted that physicians should be vigilant about taking serial patient histories and performing physical examinations because almost all soft tissue sarcomas recur within the first two years. They suggested that a chest X-ray should be taken every three months during first two years and thereafter every six months. The CT or MR imaging is useful to evaluate local recurrence and is recommended every six months. Biopsy should be done if any evidence of recurrence is detected. Accordingly, serial follow-up should be scheduled, instead of any additional treatment, in this case.
Kidney leiomyosarcoma is exceedingly rare. Because renal leiomyosarcoma does not have any diagnostic finding, it is frequently diagnosed through exploration. The best treatment of this rare disease is surgical resection rather than radiation or chemotherapy, both of which have no therapeutic advantage.9 Early detection of recurrence throughout periodic follow-up after surgical removal of the tumor is the most important treatment strategy.


 XML Download
XML Download