

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Esophageal adenocarcinomas are becoming more common.1,2 Barrett's esophagus (BE) is assumed to result from chronic esophageal reflux and is a precursor to esophageal adenocarcinoma.3-6 Reflux of duodenal contents and gastric acid in humans seems to contribute to Barrett's esophagus.7 The precise mechanism by which reflux contents cause Barrett's esophagus and predispose neoplasia is uncertain. Several recent studies, however, have shown that reflux constituents, including acid and bile, can regulate cyclooxygenase-2 (COX-2) expression.8-10 COX-2 is transiently induced by proinflammatory cytokines and growth factors and is involved in inflammation and mitogenesis.11 In addition, prostaglandin E2 (PGE2) has been shown to induce malignant change in epithelial cells through immunosuppression, inhibiting apoptosis, increasing epithelial cell metastatic potential, and promoting angiogenesis.12-16
Non-steroidal anti-inflammatory drugs (NSAIDs) may protect against cancers in the gastrointestinal tract. The protective effect is particularly well documented in the colon and rectum. For example, a 40 - 50% reduction in colon cancer incidence was reported among regular aspirin users.17 Recent studies have confirmed that regular NSAID usage is also associated with a reduced risk of stomach cancer and esophageal cancer.18-22 The molecular mechanisms underlying these chemopreventive effects are not well understood and are the subject of ongoing debate. One of the most widely accepted mechanisms for the NSAID anticancer effect concerns reduced prostaglandin synthesis due to reduced COX activity. Recent studies have reported that COX-2 inhibitors prevent esophageal adenocarcinoma in rats.23,24 Oyama et al. suggest that celecoxib is effective in preventing Barrett's esophagus and adenocarcinoma by suppressing esophagitis in rats.23
In this study, we studied the effect of sulindac on Barrett's esophagus and esophageal adenocarcinoma in surgically induced gastroduodenal reflux and COX-2 expression.
MATERIALS AND METHODS
Animals and surgical procedures
Seven-week old male Sprague-Dawley rats (Kist, Taejun, Korea) were used in this study. Throughout the experiment, all rats were housed in a controlled environment with a 12-h light/dark cycle at 22 ± 2℃. After a 1-week acclimation period, a total of 60 rats underwent operative procedures. Solid food was withdrawn for 24 h and water for 12 h before surgery. Anesthesia was induced and maintained with an isoflurane-air mixture. Esophagogastroduodenal anastomosis (EGDA) was performed.25 In short, a midline laparotomy was performed, two 1-cm incisions were made on both the gastroesophageal junction and on the anti-mesenteric border of the duodenum and were anastomosed together with accurate mucosal to mucosal opposition. Care was taken not to reach the glandular stomach when the gastroesophageal junction incision was made. Rats were allowed to drink water at 6 h post-op and were fed the following day. The Ethics committee supervising animal studies at the Dongguk University College of Medicine approved the experimental design.
Study design
Fifty-one of 60 animals operated on survived the duration of the experiment. These were divided into the control group (n = 27), the 500 ppm sulindac (Sigma, St Louis, MO, USA)-treated group (n = 15), and the 1000 ppm sulindac-treated group (n = 9). The sulindac-treated groups (n = 24) were given rat chow premixed with sulindac. An additional 12 rats underwent a sham operation and were given commercial rat chow to obtain normal esophageal tissue. Each rat was weighed every week throughout the experiment. Randomly selected rats were killed by diethyl ether inhalation on the 20th and 40th weeks after surgery. Rats were given a single intraperitoneal injection of 0.1 mg/g body weight of bromodeoxyuridine (BrdU) (Sigma) 1 h before autopsy.
Pathological analysis
The lower esophageal diameters were measured 0.5 cm away from the anastomosis site., Formalin-fixed tissues were embedded in paraffin, sectioned at 4 µm, and stained with H&E. Lower esophageal inflammation was graded as mild, moderate, or severe for histological evaluation. Barrett's esophagus was diagnosed when mucus-secreting columnar cells and goblet cells surrounded by squamous epithelium were present above the anastomosis site. Adenocarcinoma was diagnosed when glands with malignant cytological features were present both at the superficial and deep portions of the wall.
Immunohistochemistry
Serial 4-µm sections were made and spread on poly-L-lysine coated slides. Paraffin sections were immersed in three changes of xylene and hydrated using a graded alcohol series. Antigen retrieval was performed routinely by immersing the sections in 0.01 M citrate buffer (pH 6.0) and autoclaving for 15 min. Endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 15 min and incubated with monoclonal mouse anti-BrdU antibody (DakoCytomation, Carpinteria, CA, USA) overnight in a humidified chamber at 4℃ to localize proliferating cells. Staining was achieved with a DAKO LSAB + kit (Dako Cytomation) and developed with diaminobenzidine tetrahydrochloride (DakoCytomation). Sections were counterstained for 5 min with Meyer's hematoxylin and mounted. As a negative control, mouse IgG isotype (DakoCytomation) was used instead of primary antibody.
Western blot
Tissue from three rats in the same group was mixed in a tube and homogenized. Tissues were suspended in lysis buffer [10 mM Tris-HCl (pH 7.4), 1 mM EDTA, 0.25 M sucrose, 1% Triton X-100] supplemented with Complete mini protease inhibitor mixture tablets (Boehringer Mannheim, Mannheim, Germany) on ice for 1 h. After removing cell debris by centrifugation, the cell lysate protein concentration was determined by a BCA protein assay reagent (Pierce, Rockford, IL, USA) with a bovine serum albumin standard. Forty µg of protein was separated by 12% SDS-polyacrylamide gel electrophoresis and transferred to the nitrocellulose membrane. The membranes were blocked with 5% skim milk in Tris buffered saline (TBS) for 1 h at room temperature and probed with antibody overnight at 4℃. The primary antibodies were polyclonal anti-COX-2 (1 :1000, Cayman Chemical, Ann Arbor, MI, USA), anti-β-actin (1 : 200, Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse monoclonal anti-PCNA (1 : 200, Santa Cruz Biotechnology), and anti-cyclin D1 (1 : 100, Santa Cruz Biotechnology). After washing with TBS-0.05% Tween 20, the blots were treated with horseradish peroxidase-conjugated anti-rabbit IgG antibody (1 : 3000; Zymed Laboratories Inc. San Franciso, CA, USA) or anti-mouse IgG antibody (1 : 3000. Zymed Laboratories Inc.) for 1 h at room temperature. Enhanced chemiluminescence (Pierce) and autoradiography were used for detection.
Statistical analysis
We used the Kruskal-Wallis test to compare the lower esophageal diameters among the groups. When lower esophagus inflammation and Barrett's esophagus and adenocarcinoma incidences were compared, the Fisher's exact or Chi-square tests were used. Statistical significance was assumed if the p value was less than 0.05.
RESULTS
General observation
The respective number of rats examined in the control and sulindac-treated group were as follows: 8, 6, and 3 rats at week 20 and 19, 9, and 6 rats at week 40 were in the control, 500 ppm sulindac group, and 1000 ppm sulindac group, respectively. A total of 9 rats (4 control and 5 sulindac-treated) died. There was no significant difference in mortality between the two groups. Final body weights (g) were 378 ± 21 in the control, 358 ± 23 in the 500 ppm sulindac, and 392 ± 57 in the 1000 ppm sulindac at 20 weeks. They were 398 ± 40 in the control, 394 ± 48 in the 500 ppm sulindac, and 395 ± 45 in the 1000 ppm sulindac at 40 weeks. No significant difference in final body weights was found between the control and sulindac-treated groups at either time point.
Pathologic findings
The lower esophageal diameters were 10.7 ± 5.2 mm in rats undergoing EGDA and 3.8 ± 0.3 mm in sham surgery rats. Rat undergoing EGDA showed an abnormally dilated esophagus compared with sham surgery rats (p < 0.05). At 20 weeks, the control rats showed a wider lower esophagus than sulindac-treated rats, but the difference was not significant (Fig. 1A) (p > 0.05). At 40 weeks, rats treated with 1000 ppm sulindac had narrower esophageal diameters than those of the control rats (Fig. 1B) (p < 0.05). The lower esophageal mucosa displayed whitish nodular patches and superficial ulcers, which were prominent in rats with a larger lower esophageal diameter. The stomach and duodenum did not show any pathologic abnormalities.
At 20 weeks, lower esophageal inflammation was milder in the rats treated with sulindac than in the control group, but not significantly so (Fig. 2A) (p > 0.05). At 40 weeks, rats treated with 1000 ppm sulindac showed milder inflammation than control rats or those treated with 500 ppm sulindac (Fig. 2B) (p < 0.05).
Rats undergoing EGDA showed intestinal metaplasia including goblet cells above the esophagoduodenal junction (Fig. 3A) and adenocarcinoma characterized by abundant mucin secretion (Fig. 3B). At 20 weeks, Barrett's esophagus incidence was 50% in control rats and the 500 ppm sulindac-treated group and 0% in the 1000 ppm sulindac-treated group. Adenocarcinoma was present in 37.5% of the control rats and absent in sulindac-treated rats (Fig. 4A). At 40 weeks, the Barrett's esophagus incidence was similar between the control and sulindac-treated groups, but the incidence of adenocarcinoma was significantly lower in the 1000 ppm sulindac-treated group than the control or 500 ppm sulindac-treated groups (Fig. 4B) (p < 0.05). BrdU-labeled columnar cells were located in the upper and lower portion of Barrett's esophagus, while in duodenal mucosa, they were mainly restricted to the isthmic portion (Figs. 3C and D). Sulindac treatment did not change this localization (data not shown).
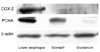
COX-2, cyclin D1, and PCNA expression
As shown in Fig. 5, COX-2 expression was significantly increased in rats exposed to gastroduodenal reflux contents for 40 weeks compared with other groups, including the sham surgery group. Moreover, its expression was localized in the lower esophagus, which also showed higher PCNA expression than the stomach and duodenum (Fig. 6). Cyclin D1 expression was increased in rats undergoing EGDA compared with those undergoing the sham surgery and with the sulindac-treatment groups (Fig. 5).
DISCUSSION
This study shows that sulindac plays a significant role in preventing the genesis of rat esophageal adenocarcinoma in surgically induced gastroduodenal reflux at least by reducing COX-2 expression.
A large body of genetic and biochemical evidence supports a role for COX-2 in human and rodent tumors.26,27 The correlation between arachidonic acid metabolism and tumorigenesis is also suggested by NSAIDs studies. Long-term NSAIDs and aspirin use protect against esophageal carcinoma.21,22 COX-2 expression was increased in human esophageal adenocarcinoma.28,29 Moreover, a selective COX-2 inhibitor suppressed rat esophageal adenocarcinoma induced by duodenal reflux.23,24,30 In this study, sulindac treatment also reduced esophageal adenocarcinoma development. COX-2 expression was also increased in Barrett's mucosa in response to acid or bile acid pulses in an ex vivo organ culture system. This effect was attenuated by a selective COX-2 inhibitor.9 The effect of a COX-2 inhibitor in preventing the genesis of Barrett's esophagus in animal studies is controversial. Buttar et al. did not observe any suppressive action of COX-2 inhibitors on the development of Barrett's esophagus.24 In contrast, a recent study reported that a COX-2 inhibitor prevented the esophageal inflammation-Barrett's esophagus-adenocarcinoma sequence in rats.23 The present study reveals that the incidence of Barrett's esophagus is lower only in rats treated with 1000 ppm sulindac at 20 weeks. This means that the genesis of Barrett's esophagus is dependent on reflux duration and COX-2 inhibitor dose.
Increased proliferating cells and an expanded proliferative compartment were demonstrated in Barrett's esophagus and adenocarcinoma.31,32 Therefore, we studied the localization of BrdU-labeled cells and cyclin D1 expression. In this study, BrdU-labeled columnar cells were located in the upper and lower portions of Barrett's esophagus, while they were mainly restricted to the isthmic portion in duodenal mucosa. This study showed that cyclin D1 and PCNA expression were increased in the lower esophagus of rats exposed to gastroduodenal reflux contents. In addition, sulindac treatment reduced cyclin D1 expression. Increased nuclear cyclin D1 expression was observed in 22 - 64% of the esophageal adenocarcinomas.33 Recent studies report that NSAIDs suppress cancer cell growth by inhibiting cyclin D1 expression.34,35
In conclusion, we suggest that the chemopreventive effect of sulindac is related to decreased COX-2 and cyclin D1 expression, which may be influenced by reduced inflammation.





 XML Download
XML Download