

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Toxoplasma gondii (T. gondii) is an obligate intracellular protozoan parasite that causes congenital infections in newborns and opportunistic infections in immunocompromised patients, such as those with AIDS. T. gondii infects all nucleated cells, eventually causing the host cell to rupture following replication of the tachyzoite.
Cell survival or death, as induced by apoptosis, is a physiological response to cell damage caused by external and internal stimuli, and the exact fate of a cell depends on the balance of pro-apoptotic and anti-apoptotic proteins. Apoptosis limits the growth of infectious agents and plays a crucial role in the immune response. There are two pathways of apoptosis of cells: one triggered by death signals, and another involving the mitochondria. Activation of death receptors triggers the cleavage of caspase 8, whereas release of cytochrome c from mitochondria activates caspase 9; both pathways eventually converge to activate the execution caspase, caspase 3.1 While the pathogens Acanthamoeba sp. and E. histolytica can induce host cell apoptosis,2-4 the anti- or pro-apoptotic activities associated with T. gondii remain controversial. Induction of apoptosis in Peyer's patch T cells and the ocular tissue of mice infected with T. gondii has been reported.5,6 The Fas/Fas L pathway may play a crucial role in the induction of IFN-γ mediated apoptosis in N. caninum and T. gondii infected cells,7 and infection with T. gondii (RH strain) results in an over production of IFN-γ and high levels of apoptosis.8 Conversely, some intracellular protozoa have the ability to inhibit host cell apoptotic responses. T. gondii activates a survival response in infected cells by increasing host resistance to multiple apoptotic stimuli.9 In the present study, we focus on the inhibition of apoptosis in mouse spleen cells infected with T. gondii using morphological, molecular, and biochemical approaches to characterize the role of NF-κB in the anti-apoptotic response.
MATERIALS AND METHODS
Toxoplasma gondii
T. gondii tachyzoites (RH) were maintained in the peritoneal cavity of ICR mice (Dae-Han BioLink Co., Eumseong-gun, Chungcheongbuk-do, Korea) for three to four days. Tachyzoites were harvested in RPMI 1640 medium (Invitrogen, Carlsbad, CA, USA) from the peritoneal cavity and separated from peritoneal cells by repeated low and high speed centrifugation (40 × g for 5 min and 650 × g for 10 min).
Treatment of mouse spleen cells with T. gondii infection and actinomycin-D
Spleen cells in RPMI 1640 media were harvested from BALB/c mice and pelleted by centrifugation (650 × g for 10 min). Cells were resuspended in lysis buffer (0.83% NH4Cl-0.01M Tris-HCl) in order to remove red blood cells. Mouse spleen cells (5 × 106 cells/well) were cultured in 6-well plates with RPMI 1640 media containing 2 mM L-glutamine, 2 mg/mL NaHCO3, 10% fetal bovine serum (FBS, Cambrex, Walkersville, MD, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin at 37℃, 5% CO2 atmosphere. Cells were infected with 5 × 107 T. gondii tachyzoites and/or treated with 5 - 10 µg/mL actinomycin-D (AD, Sigma-Aldrich, St. Louis, MO, USA) for 5 h and 18 h, respectively.
DNA size analysis by agarose gel electrophoresis
Infected and non-infected mouse spleen cells were harvested by centrifugation and resuspended in NET buffer containing 10% SDS, DNA-free Rnase, and proteinase K. The cells were then centrifuged at 12,000 × g for 5 min at 4℃, and DNA was extracted from the cell pellet with phenol, phenol/isoamyl alcohol, and chloroform, followed by precipitation with 100% ethanol and 10 M NH4OAc. The DNA was dissolved in TE buffer and quantified by A260 on a spectrophotometer (Perkin Elmer, Wellesley, MA, USA). DNA samples were analyzed on a 2% agarose gel stained with 1 µg/mL ethidium bromide.
Flow cytometry
Cells were stained with annexin V using the Annexin V/PI kit (R&D systems, Minneapolis, MN, USA), and apoptotic cells identified and quantified by flow cytometry. Briefly, spleen cells (1 × 106) were washed in PBS and incubated with 10 X binding buffer, propidium iodide (PI), annexin V-FITC, and dH2O (total 100 µL) for 15 min at room temperature. The apoptotic cells were analyzed on a fluorescence-activated cell sorter (FACS, Becton Dickinson-FACScalibur, Cockeysville, MD, USA).
Reverse transcriptase (RT)-PCR
Bcl-2 family messenger RNA was identified by RT-PCR using message specific primers. Mouse spleen cells were incubated with AD and AD/T. gondii for 30 min, 60 min or 120 min, and total cellular RNA extracted using TRIzol (Invitrogen), chloroform, and isopropyl alcohol. Reverse transcription was performed with an oligo dT primer, and cDNAs were amplified by PCR using the primers listed in Table 1. PCR products were resolved on a 2% agarose gel stained with 1 µg/mL ethidium bromide.
Western blot analysis of Bcl-2 family proteins and caspases
The abundance of Bcl-2 family proteins (Bax, Bcl-2, Bcl-XL), caspase 3, caspase 8, PARP and actin in mouse spleen cells was analyzed by Western blotting. Spleen cells (5 × 105) were resuspended in lysis buffer (20 mM Tris-HCl, PH 7.5, 60 mM beta-glycerophosphate, 10 mM EDTA, 10 mM MgCl2, 10 mM NaF, 2 mM DTT, 1 mM Na3VO4, 1 mM APMSF, 1% Np-40, 5 µg/mL leupeptin). Following a 40 min incubation on ice, the lysates were combined with 2 X sample buffer (0.125 M Tris-HCl, pH 6.8, 4% SDS, 20% glycerol, 10% 2-mercaptoethanol, 0.2% bromphenol blue) and boiled for 5 min, samples were resolved on a SDS-polyacrylamide gel. After semi-dry transfer to a PVDF membrane (Amersham Pharmacia Biotech, Uppsala, Sweden), ponceau S staining was used to confirm loading of equal amounts of total protein loaded in each lane. The membranes were blocked (5% nonfat dried milk, 0.1% Tween 20, 100 mM NaCl, 10 mM Tris-HCl, PH 7.6) for 1 h and incubated with the appropriate primary antibody: monoclonal mouse anti-Bax antibody (BD PharMingen, San Diego, CA, USA, 2 µg/mL), polyclonal rabbit anti-Bak antibody (BD Phar Mingen, 2 µg/mL), monoclonal mouse anti-Bcl-2 antibody (Oncogene, San Diego, CA, USA, 2 µg/mL), polyclonal rabbit anti-caspase 3 antibody (Cell Signaling, Beverly, MA, USA, 1:100), monoclonal mouse anti-poly (ADP-ribose) polymerase (PARP) antibody (BD Phar Mingen, 1:200), polyclonal rabbit anti-caspase 8 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA, 1:200), or polyclonal rabbit anti-actin antibody (Santa Cruz Biotechnology, 1:200). Bound primary antibodies were visualized with an ECL anti-mouse IgG peroxidase-linked species-specific F(ab')2 fragment (Amersham Pharmacia Biotech, 1:10,000) or an ECL anti-rabbit IgG peroxidase-linked species-specific F(ab')2 fragment (1:200,000) and the ECL PLUS chemiluminescence detection system employed according to the manufacture's instructions (Amersham Pharmacia Biotech).
Western blot for NF-κB and IκBα
Nuclear and cytoplasmic fractions were prepared from spleen cells.10 Monoclonal mouse IκBα antibody (Cell signaling, 1:100), rabbit phospho-NF-κB p65 antibody (Cell signaling, 1:50), NF-κB p65 antibody (Cell signaling, 1:100) and mouse anti-actin antibody (Chemicon international, Temecula, CA, USA, 1:1,000) were used for detection of NF-κB and IκBα by Western blot as described above.11
Electrophoresis motility shift assay (EMSA)
Nuclear extracts from spleen cells were prepared as previously described.10 Protein concentrations of the nuclear extracts were determined by Bradford assay, and EMSAs were performed according to the manufacture's instructions (Promega, Madison, WI, USA). Briefly, 5 µg of nuclear extract was incubated with a (γ32P)-labeled oligonucleotide probe corresponding to the consensus NF-κB binding site for 30 min at room temperature. After incubation, bound and free DNAs were resolved on a 5% polyacrylamide gel12 run at 100 V for 3 h, dried, and subjected to autoradiography for 12 h to 48 h.13
Transmission electron microscopy (TEM)
The apoptotic changes of mouse spleen cells treated with AD (10 µg/mL) and/or infected with T. gondii (3 × 106, cell:tachyzoite = 1:10) for 6 h in 6 well plates were visualized by TEM performed using standard methods. Briefly, spleen cells were fixed in 3% glutaraldehyde (PH 7.8) for 3 h at room temperature and post-fixed in 1% osmium tetroxide for 1.5 h at 4℃. After serial dehydration with 50%, 70% and 100% ethanol, specimens were embedded in Epon resin. Ultrathin sections were loaded onto copper grids; stained with uranyl acetate and lead citrate, and visualized with a transmission electron microscope H-7600s (Hitachi, Tokyo, Japan).
RESULTS
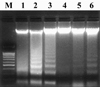
DNA fragmentation in spleen cells was suppressed by T. gondii
Spleen cells were incubated with AD and/or T. gondii tachyzoites for 5 h and 18 h respectively at 37℃. DNA was then extracted from spleen cells, resolved on a 2% agarose gel, and stained with ethidium bromide. DNA laddering was observed in mouse spleen cells treated with AD for 5 h (data not shown) and 18 h (Fig. 1, lanes 1 & 2). Apoptosis was down regulated in mouse spleen cells treated with AD and infected with T. gondii (Fig. 1, lane 6). Spleen cells infected with T. gondii only showed weak DNA fragmentation (Fig. 1, lane 5). DNA laddering was observed in spleen cells for 18h incubation, but not in freshly isolated cells (Fig. 1, lanes 3 & 4). Similar results were found upon repeated experiments.
Inhibition of apoptosis in spleen cells infected with T. gondii was observed by flow cytometric analysis
Mouse spleen cells treated with AD and/or T. gondii infection were pooled, incubated with annexin-FITC V and PI, and analyzed by flow cytometry. Early apoptotic cells have increased annexin V, but not PI staining. Late apoptotic or necrotic cells stain with both annexin V and PI. Mouse spleen cells cultured with AD, AD/T. gondii and freshly isolated cells showed apoptosis of 43.6%, 37.0%, and 15.8%, respectively after incubation for 5 h (data not shown). The quantity of apoptotic spleen cells were 80.3%, 30.1%, and 23.8%, respectively, after incubation for 18 h (Fig. 2, A, D, F). Similar results were found upon repeated experiments.
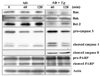
Levels of Bax mRNA and protein expression in spleen cells were reduced upon treatment with T. gondii
RT-PCR analysis of Bcl-2 family genes was performed on mouse spleen cells incubated with AD, or AD/T. gondii for 30 min, 60 min or 120 min at 37℃. Expression of pro-apoptotic Bax mRNA was down regulated in AD/T. gondii infected spleen cells (Fig. 3). Western blot analysis showed that the production of Bax protein was decreased in spleen cells treated with AD and infected with T. gondii, while Bak protein levels remained unchanged (Fig. 4). Expression levels of anti-apoptotic Bcl-2 and Bcl-XL mRNAs remained constant in spleen cells treated with AD and infected with T. gondii (Fig. 3). Spleen cells infected with T. gondii showed indistinct Bax and Bcl-2 mRNA levels. Moreover, Bcl-2 protein did not appear in AD/T. gondii infected cells.
Caspase 3 and PARP protein activation was suppressed in T. gondii infected spleen cells
Activation of caspase 3 and PARP proteins was suppressed in spleen cells treated with AD and infected with T. gondii. Down regulated, cleaved caspase 3 at 120 min post infection and reduced production of PARP in T. gondii infected cells were detected by Western blotting (Fig. 4). Although cleaved caspase 8 was observed in spleen cells treated with AD and infected with T. gondii, it was not expressed in spleen cells treated with AD only (Fig. 4).
Activation of NF-κB in spleen cells infected with T. gondii
We examined the role of NF-κB in the anti-apoptotic response of T. gondii infected spleen cells by EMSA and Western blotting. NF-κB activity was apparent at 120 min in spleen cells treated with AD and infected with T. gondii, which decreased by 180 min (Fig. 5A). T. gondii infected spleen cells showed more activated NF-κB than uninfected cells at 60 min on EMSA. A similar pattern of NF-κB activity in total cell and nuclear extracts was observed by Western blot (Fig. 5B). Decreased IκBα was associated with increased NF-κB activity.
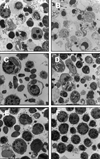
TEM shows inhibition of apoptosis in T. gondii infected spleen cells
Condensation of nuclear chromatin and nuclear fragmentation was observed in spleen cells treated with AD (Fig. 6A & B); however, these two events were not observed in spleen cells treated with AD and infected with T. gondii tachyzoites (Fig. 6C & D). In spleen cells infected with T. gondii only, neither nuclear condensation nor fragmentation was observed (Fig. 6E).
DISCUSSION
Apoptosis, a mechanism of programmed cell death, is a result of both physical and pathological events. Transduction of pro-apoptotic stimuli via signaling pathways results in the activation of the caspase family of cysteine proteases. Bcl-2 family pro-apoptotic proteins promote cell death by inducing mitochondrial damage, cytochrome c release, and subsequent caspase activation, and the Bcl-2 family of anti-apoptotic proteins inhibit apoptosis by preventing the release of cytochrome c; however, the mechanism by which parasites utilize this method of apoptotic inhibition has not been elucidated. Down-regulation of host cell apoptosis may also provide an explanation for the persistence of intracellular pathogens. T. gondii has several strategies for inhibiting the initiation of the apoptotic cascade. In the present study, we found that apoptosis was not induced in mouse spleen cells infected with T. gondii when compared to uninfected controls (data not shown); however, apoptosis was induced in spleen cells by treatment with AD only. One biochemical analysis of apoptosis activity is the presence of oligonucleosome-sized fragments of DNA, which produce ladders when analyzed with agarose gel electrophoresis. Our results indicated that the induction of DNA fragmentation in AD treated spleen cells was inhibited by T. gondii infection (Fig. 1). Apoptotic cells can be detected by the binding of fluorescent labeled annexin V to the plasma membrane. As such, early and late apoptotic cells were measured by flow cytometric analysis after double staining with FITC-conjugated annexin V and PI.14 While flow cytometric analysis could not differentiate late apoptosis and necrosis of cells, it demonstrated that concomitant infection with T. gondii abrogated AD induced apoptosis. Specifically, in the AD treated population, 80.3% of cells were apoptotic, as opposed to 30.1% of the cells treated with both AD and T. gondii after incubation for 18 h (Fig. 2). Proteins of the Bcl-2 family with pro-apoptosis or anti-apoptotic functions reside in the outer mitochondrial membrane. Bax, a pro-apoptotic protein, triggers the release of cytochrome c from mitochondria.1 Conversely, Bcl-2 and Bcl-XL are capable of blocking the release of cytochrome c, thereby preventing the activation of caspase 9. The induction of apoptosis by T. vaginalis is associated with a reduction of expression of anti-apoptotic Bcl-XL protein, but not Bcl-2, and the activation of caspase 9 suggests that mitochondria play a role in T. vaginalis-induced RAW 264.7 programmed cell death.13 RT-PCR analysis demonstrated that T. gondii infection promotes the activation of anti-apoptotic Bcl-2 family members but not pro-apoptotic pathways.15 Our studies showed that the expression of Bax mRNA and protein was indistinct in AD/T. gondii-treated mouse spleen cells, compared to uninfected cells (Fig. 3). Likewise, expression of Bcl-2 and Bcl-XL mRNA and Bak protein were unchanged in mouse spleen cells treated with AD and infected with T. gondii (Figs. 3 & 4). These results suggested that the anti-apoptotic Bcl-2 protein was not affected in T. gondii infection and that the other anti-apoptotic protein, Bcl-XL may be involved. Generally T. gondii infection promotes the suppression of pro-apoptotic Bax, but not anti-apoptotic Bcl-2 proteins (Fig. 4).
Caspase 3 is a critical downstream member of the caspase family, and is an essential effector of apoptosis. The resistance of T. gondii infected cells to apoptosis may be attributed to prevention of caspase 3 activity.1,7,16 In the present study, inactivation of caspase 3 at 120 min post infection and decreased PARP were present in cells treated AD and infected with T. gondii as determined by Western blotting (Fig. 4). Cleaved caspase 8, which was present in cells treated with AD and T. gondii infection, may have induced a weak apoptosis response. We propose that AD did not trigger apoptosis via the caspase 8 pathway and that infection by the parasite induced apoptosis slightly. Activation of the caspase cascade leads to cleavage of a variety of target proteins including PARP, nuclear lamins, and protein kinase C (PKC); both PARP protein levels and cleavage of nuclear target proteins are down regulated by T. gondii.16 Our results showed that T. gondii mediates inhibition of apoptosis by a blockade of Bax expression and caspase 3 activation. Inhibition of apoptosis by T. gondii is regulated at the transcriptional level of NF-κB, as infection with T. gondii induces the activation of NF-κB and the transcription of anti-apoptotic genes.17-21 Phosphorylation of IκB activates and promotes NF-κB translocation to the nucleus, where NF-κB associates with the transcription of proinflammatory mediators. Further, expression of NF-κB is correlated with anti-apoptotic protein expression after T. gondii infection. In this experiment, spleen cells treated with AD and infected with T. gondii showed increased NF-κB DNA binding activity by EMSA (Fig. 5). Condensation of chromatin, disruption of nuclear architecture, vacuolation of the cytoplasm, membrane blebbing, detachment from substrate, and cell shrinkage are some of the morphological changes associated with apoptosis. We observed some of these morphologic changes in cells treated with AD and/or T. gondii using TEM. Condensed chromatin and fragmented nuclei of cells were apparent 6 h after treatment of the cells with AD; however, there were no apoptotic changes in cells treated with AD and T. gondii (Fig. 6C & D). In contrast to the inhibition of apoptosis in T. gondii infected cells, parasite infection induced apoptosis has been reported in neighboring non-invaded cells.5,6,22 However, we were unable to identify such changes in uninfected mouse spleen cells on TEM (Fig. 6D & E).
In conclusion, infection with T. gondii induces an anti-apoptotic effect, which was associated with inactivation of caspase 3 and PARP. Host cell mitochondria around the parasitophorous vacuole may be involved in anti-apoptotic activity by T. gondii.18 To survive in host cells, parasitic protozoa modulate their host's apoptotic response. These findings suggest that mouse spleen cells infected with T. gondii resisted AD treatment, an apoptotic stimuli, by inactivating the caspase cascade and suppressing Bax protein expression through NF-κB activation. These results provide direction for further investigation of the mechanism of inhibition of apoptosis by T. gondii in host cells.




 XML Download
XML Download