

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Toluene diisocyanate (TDI) is one of the most commonly identified causes of occupational asthma in far-east Asia, with a prevalence of 2.9-13% in exposed workers.1,2 The pathogenesis of TDI-induced asthma remains incompletely understood, and reliable methods of serological testing have not been established.
Long-term follow-up studies have shown that more than 50% of patients with TDI-induced asthma have persistent asthmatic symptoms after complete avoidance of exposure and after taking anti-asthmatic medications.3 Early diagnosis is the best way to prevent TDI-induced asthma. There have been several studies finding serologic markers for early diagnosis. However, the prevalence of anti-TDI-HSA (human serum albumin) antibodies was 0-50%, and the sensitivity of anti-TDI-HSA antibodies was less than fifty.4 More studies are needed to determine standardized methods for detecting specific antibodies. Recent investigations have demonstrated that inhaled hexamethylene diisocyanate could conjugate to human airway epithelial keratins to initiate an immune response.5 Also, our recent study showed that TDI exposure could increase expression of cytokeratin (CK) 19 in human bronchial epithelial cells of patients with TDI-induced asthma.6 In this study, we extended these findings and evaluated the clinical significance of autoantibodies to three major epithelial CKs - CK8, 18, and 19 - in serum samples from patients with TDI-induced asthma, as compared with exposed and unexposed control subjects.
MATERIALS AND METHODS
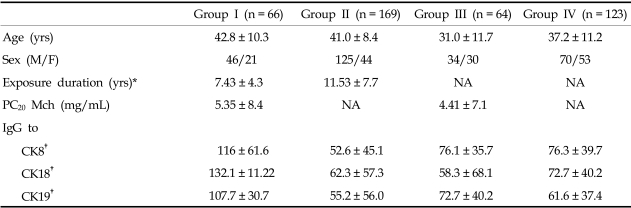
Sixty-six patients with TDI-induced asthma, confirmed by positive responses to TDI bronchoprovocation, were enrolled and classified as group I. 169 exposed asymptomatic workers (group II), 64 allergic asthma patients that were sensitive to house dust-mite allergens (group III), and 123 unexposed healthy subjects (group IV) were enrolled as control subjects. Atopy was determined by a positive skin test to at least one common inhalant allergen, including house dust mites, tree and pollen mixtures, mugwort pollen, and ragweed pollen, Alternaria (Bencard, Brentford, U.K.). The demographic data of the four study groups are compared in Table 1. Serum samples from group I and III subjects were collected before treatment; all subjects stopped using inhaled or oral steroids four weeks before the study. Subjects underwent an interview, chest radiography, skin-prick test with common inhalant allergens, lung function measurement, and inhalation challenge with methacholine. All subjects gave informed consent, regulated by the Institutional Review of Board of Ajou Medical Center, Suwon, Korea.
Bronchial challenge test with methacholine and TDI
Subjects in groups I and III underwent methacholine bronchial challenge, according to methods previously described.4 Briefly, aerosols were generated by a DeVilbiss 646 nebulizer, connected to a Devilbiss dosimeter, driven by compressed air (Devilbis Co., Doylestown, PA, USA). Five inhalations of normal saline at 5-min intervals were administered, followed by a series of successively doubled doses of methacholine (0.075-25 mg/mL) until a 20% decrease in FEV1 was observed or the maximum dose was given. FEV1 was measured 5 min after the beginning of each set of inhalations of aerosolized methacholine. The methacholine PC20 level was determined by interpolation from the dose-response curve. The TDI bronchial challenge test was performed, according to a previously described protocol.4
Preparation of the TDI-HSA conjugate
2,4-TDI-HSA conjugates were prepared, as described previously, by using a modified version of Tse and Pesce's method.4 2,4-TDI (2.4g) was added to 90 mL of 1% HSA in PBS with constant stirring. Aliquots were taken 5, 10, 20, 30, and 40 min after the reaction. Ten milliliters of 1% HSA solution was used as an unconjugated control. Ammonium carbonate (2 M) was added to each aliquot to terminate the reactions. All reactive samples were centrifuged at 3,000 g for 40 min at 4℃ to remove unreactive TDI. They were extensively dialyzed for three days against 0.1 mol/L ammonium carbonate and precipitated with an equal volume of 20% trichloroacetic acid. Samples were then redissolved in 1 mol/L sodium hydroxide and dialyzed with 4 L of deionized water for one day. When the degree of substitution was determined by a modified Gutmann assay, the molar ratio was 5. The protein content of a conjugated sample was determined by the Lowery method. All reagents used for the preparation of TDI-HSA conjugates were purchased from Sigma Chemical Co. (St Louis, MO, USA).
ELISAs to measure anti-TDI-HAS IgG and IgE antibodies
The serum IgG level was detected by ELISA, as described previously.4 In brief, after 1 µg of TDI-HSA conjugate or mock conjugate was dissolved in sodium bicarbonate buffer (pH 9.5), it was coated onto an ELISA plate individually (Corning, New York, NY, USA) at 4℃. After washing with PBS-Tween 20 (PBS-T), 350 µL of blocking buffer (PBS that contained 1% BSA and 0.1% Tween 20) was added. Serum from patient or control subjects was diluted 1 : 5 for IgG and 1 : 3 for IgE in the preliminary experiments. 50 µL of diluted serum was incubated for 1 hr at 37℃ in both TDI-HSA- and HSA-coated wells, which were then washed three times. Peroxidase-conjugated anti-IgG (1 : 2000 v/v, 50 µL; Sigma) or biotin-conjugated anti-IgE antibody (1 : 200 v/v, Sigma) was added to each well, incubated for 1 hr at room temperature, and washed with PBST. 100 µL of streptavidin-peroxidase was added to each IgE-ELISA well for 30 min of incubation at 37℃. Then, 100 µL of substrate solution, dissolved in substrate buffer (3,3', 5,5' tetramethylbenzidine, Sigma), was added. After 15 min of incubation at 37℃, the reactions were stopped with the addition of 2.5 N H2SO4. Absorbance values were read, using an ELISA reader at 405/450 nm. The final absorbance of each sample was normalized by subtracting the HSA-coated value from the TDI-HSA-coated value. The positive cutoff value (0.18 for IgG, and 0.13 for IgE) was determined to be the mean absorbance of 123 unexposed healthy control subjects plus 2 standard deviations (SD).
IgG autoantibodies to three CKs by ELISA
Serum-specific IgG levels were detected by ELISA using 50 µL of diluted (1 : 20) patient serum or negative control serum. This was determined to be the optimal concentration in the preliminary experiment. Prior to the addition of the serum, each well of the ELISA plates was coated with 100 ng/well of CK8, 18, and 19 (Research Diagnostics, Flanders, NJ, USA), dissolved in PBS, and was blocked with 200 µL of blocking buffer (PBS that contained 10% FBS). After 1 hour of incubation at room temperature, the plate was washed three times with PBS-T. Alkaline phosphatase-conjugated anti-human IgG (100 µL; Sigma), diluted to 1 : 10000 v/v with 10% FBS-PBS, was incubated for 1 hour at 37℃, and was washed with PBS-T. PNPP (p-nitro-phenyl phosphate; Sigma) was added as the substrate. Reactions were stopped with the addition of 1 N NaOH. The optical density of the solution was determined at 405 nm by using an ELISA reader. The final absorbance was determined by subtracting the absorbance of the uncoated well. Each absorbance was presented in arbitrary units, based on the standard curve, which was derived from serial dilutions of the pooled serum samples with high levels of CK8, CK18, and CK19 autoantibodies. The positive cutoff value was determined as the mean absorbance of 123 unexposed healthy control subjects plus 2 SD.
ELISA inhibition test
Increasing amounts (0.1-100 µg/mL) of each CK were dissolved in the blocking solution (PBS that contained 0.005% Tween 20 and 0.5% nonfat dry milk) and incubated for 1 hour with pooled serum samples from patients with TDI-induced asthma, who had high IgG levels. Then the CK-added samples were added to the wells of the antigen-coated microtiter plates, and ELISA experiments were repeated as described above. The percentage of inhibition was calculated as follows: % inhibition = 100 × (1-absorbance with inhibitor)/(absorbance without inhibitor).
CK immunoblots
Recombinant cytokeratins in 30 mM TRIS hydrochloride, 2 mM EDTA, 9.5 M urea, 2 mM DTT, and 10 mM methylammonium chloride (pH 8.0) were separated by discontinuous SDS-polyacrylamide gel electrophoresis (PAGE), using a 4-20% Tris-glycine gradient gel (Invitrogen, Carlsbad, CA, USA). After electrophoresis, proteins were transferred onto a polyvinylidene difluoride membrane (Millipore, Billerica, Mass, USA) and blocked with 5% nonfat milk. After the transfer, membrane strips were probed with monoclonal antibody to CK8, CK18, and CK19 (Sigma) at a 1 : 20 dilution for 3 hours at room temperature. After washing, each membrane strip was incubated with alkaline phosphatase-conjugated goat anti-human IgG (Sigma) for 1 hour at room temperature. The membrane strips were washed again and then stained with nitroblue tetrazolium/5-bromo-4-chloro-3-indoyl phosphate (Sigma). The intensity was measured using a densitometer and presented as the relative intensity.
Measurement of antinuclear antibody (ANA)
ANA autoantibodies were detected, using Hep Scan ANA in the serum samples from patients with TDI-induced asthma and diagnostic kits (BL Diagnostika, Hamburg, Germany), according to manufacturer's instructions.
Statistical analysis
The ANOVA test, followed by a post hoc Scheffe test, was applied to compare demographic data and the mean level of CK8, CK18, and CK19 autoantibodies among the four study groups. The Student's t-test was applied to compare PC20 methacholine dose, according to the presence of specific CK autoantibodies. These continuous variables did not have a normal distribution and were log-transformed. They were presented in their original scale. The cross-tab analysis was applied to compare the prevalence of specific CK autoantibodies with specific antibodies to TDI-HSA conjugate or with laboratory findings. A p-value of 0.05 or less was regarded as statistically significant.
RESULTS
CK8, 18, and 19 autoantibodies
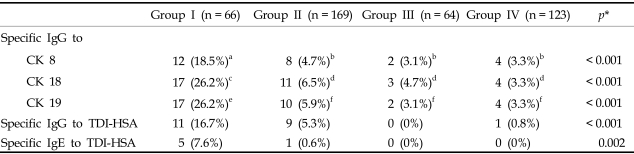
Fig. 1 shows the IgG levels for CK8 (Fig. 1A), CK18 (Fig. 1B), and CK19 (Fig. 1C), obtained by ELISA, in the four study groups. When the autoantibody levels for the three CKs were compared, there were significant differences among the four groups. Autoantibody levels to the three CKs were significantly higher in group I than in the three control groups (p < 0.05, respectively; Table 1). In group I, CK18 and CK19 (26.2% each) autoantibodies were more prevalent than those of CK8 (18.5%). These values were significantly higher than those of groups II, III, and IV (p < 0.05, Table 2).
Association of anti-TDI-HSA antibodies and clinical parameters
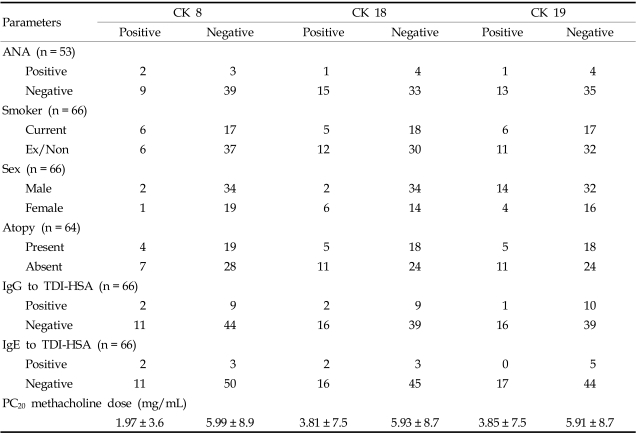
The prevalence of CK autoantibodies was not significantly associated with that of anti-TDI-HSA antibodies and clinical parameters such as sex, atopy, smoking status, and ANA in patients with TDI-induced asthma (p > 0.05; Tables 3, 4). In group I, the PC20 methacholine doses were low in patients with CK autoantibodies, although the significances were not documented (for IgG to CK8 1.97 ± 3.6 vs 5.99 ± 8.9, p = 0.051, for IgG to CK18 3.81 ± 7.5 vs 5.93 ± 8.7, p > 0.05, for IgG to CK19 3.85 ± 7.5 vs 5.91 ± 8.7, p > 0.05, Table 3). Among the subjects in group I, a significant association was noted between the prevalence of CK19 autoantibodies and the prevalence of CK8 and CK18 autoantibodies (p < 0.001; data not shown).
ELISA inhibition tests for the three CKs
Fig. 2 shows the results of the ELISA inhibition test for subjects that tested positive for CK autoantibodies. Significant, dose-dependent inhibition was noted for each CK.
CK immunoblots
Fig. 3 shows an immunoblot analysis of the three CKs. Serum samples from patients with TDI-induced asthma, unexposed healthy control subjects, and buffer controls were compared with monoclonal antibodies to the three CKs. Specific binding to the three CKs was confirmed.
Sensitivity and specificity of CK autoantibodies in TDI-exposed workers
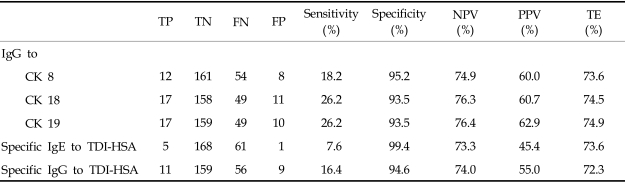
Table 4 demonstrated the sensitivity, specificity, test efficiency, and positive and negative predictive values of specific CK autoantibodies and anti-TDI-HSA antibodies in the study subjects. The sensitivity and positive predictive value of CK18 and CK19 autoantibodies were higher than those of anti-TDI-HSA antibodies. Specificity, negative predictive value, and test efficiency were similar in these two groups.
DISCUSSION
In this study, higher autoantibody levels for the three epithelial CKs (CK8, CK18, and CK19) were detected in serum samples from patients with TDI-induced asthma than in asymptomatic exposed workers, in patients with allergic asthma and healthy unexposed control subjects. Among the three autoantibodies, those to CK18 and CK19 showed the highest sensitivities. Furthermore, the prevalence CK18 and CK19 autoantibodies were significantly higher in patients with TDI-induced asthma than in asymptomatic exposed workers, and their binding specificities were confirmed by ELISA inhibition tests. Based on the prevalence of anti-TDI-HSA antibodies observed in this study, we suggest that serum CK18 and CK19 autoantibodies might be used as serological markers for identifying patients with TDI-induced asthma among exposed workers, although the sensitivity was not high enough (26.2%).
Previous studies on the long-term prognosis of TDI-induced asthma suggest that early diagnosis of sensitized patients and immediate removal from the exposure could increase the likelihood of remission. However, an appreciable number of patients with TDI-induced asthma do not recover completely, even after avoidance and treatment.3,4,7 Therefore, developing an early diagnostic marker based on serologic tests is essential in TDI-induced asthma. Although monocyte chemoattractant protein-1 (MCP-1) could be a sensitive test to identify diisocyanate (TDI, MDI, or HDI) induced asthma,8 this is not easy to repeat in a larger sample. Several investigators have detected anti-TDI-HSA IgE and IgG antibodies in serum samples from patients with TDI-induced asthma, using ELISA or RAST. The prevalence of anti-TDI-HSA IgE antibodies has proven quite variable between laboratories, and sensitivity to IgG was not high enough.4,9 This study is the first to evaluate whether autoantibodies to major bronchial epithelial CKs can be applied to the identification of asthma patients exposed to a single type of isocyanate (patients with TDI-induced asthma vs. TDI-exposed, but asymptomatic workers), and sensitivity, specificity, positive and negative predictive value were compared with those of anti-TDI-HSA IgE and IgG antibodies. The sensitivities and positive predictive values of CK18 and CK19 autoantibodies were higher than those of anti-TDI-HSA IgE and IgG antibodies, while the specificities, negative predictive values, and test efficiencies were similar. Significant associations were not found between the prevalence of specific IgE and IgG antibodies. These findings suggest that autoantibodies to CK18 and CK19 may be used as a supplementary serologic markers to increase the diagnostic sensitivities of serum-specific antibody tests.
Several airway epithelial cell proteins can conjugate with diisocynates in vitro and in vivo.6,10-12 CKs, major intracytoplasmic cytoskeleton proteins, are potential self-antigens, with a total of 20 human epithelial keratins, based on molecular weight and isoelectric point. Pairs of CKs seem to be consistently coexpressed in different types of epithelial cells.13 The three CKs applied in this study have been found in both bronchial and lung alveolar epithelial cells, which are the major target tissues of bronchial asthma. The prevalence of specific CK18 and CK19 autoantibodies were the same with similar inhibition potencies, according to the ELISA inhibition test.
CKs are normally located in the intracellular space and could gain access to the immune system after epithelial damage or cell death. The precise mechanism of their generation is still unknown, although several studies suggest that CKs are proteolyzed during cell apoptosis and can leak into the circulation as a soluble form, where they may serve as new epitopes for antibody generation.14-17 In this study, high levels of specific CK autoantibodies were detected only in patients with TDI-induced asthma, not in those with allergic asthma. Therefore, these findings may not be derived from simple epithelial damage found in asthmatic airways. Among the patients with TDI-induced asthma, those who are more susceptible to epithelial damage by TDI exposure may develop serum-specific CK autoantibodies. In this study, because we found no association between these antibodies and the clinical characteristics of TDI-induced asthma, including asthma symptoms and duration of exposure, we could not speculate as to which patients would be susceptible. However, airway hyper-responsiveness to methacholine was more severe in patients with autoantibodies to any of the three CKs, despite marginal statistical significance.
Further studies are needed to investigate genetically susceptible markers for developing autoantibodies to specific CKs among workers exposed to TDI. How diisocyantes and human airway epithelial cells are involved in airway inflammation has not been studied fully.18 HDI, conjugated with bronchial CK18, can induce T-cell proliferation and cytokine production in patients with diisocyanate asthma.9,18 Further studies are needed to confirm whether the development of autoantibodies to specific CKs is an epiphenomenon of chronic airway damage occurring in patients with TDI-induced asthma or whether pathogenic or autoantibody-mediated mechanisms are involved.
In conclusion, we demonstrated circulating autoantibodies to three bronchial epithelial CKs (8, 18, and 19) in patients with TDI-induced asthma. Among these, autoantibodies to CK18 and 19 may be used as serological markers for the detection of TDI-induced asthma among workers with high-risk exposure. Further studies will be needed to investigate how these specific autoantibodies may be involved in the chronic, persistent airway inflammation found in TDI-induced asthma.



 XML Download
XML Download